
Изо расшифровка: Как расшифровывается изо? Значения аббревиатур и сокращений на сайте klmn.price-review.ru
ISO | ИСО – Международная организация по стандартизации в вопросах и ответах | Что такое ИСО?
Что такое ИСО?
Международная организация по стандартизации (ИСО) является одной из самых крупных и значимых организаций, занимающейся разработкой международных стандартов.
Что такое стандарт ИСО?
Стандарт ISO – документ, устанавливающий требования, спецификации, руководящие принципы или характеристики, в соответствии с которыми могут использоваться материалы, продукты, процессы и услуги, которые подходят для этих целей.
Обязательно ли применение стандартов ИСО в деятельности организации?
Стандарты ИСО являются добровольными, при этом страны могут принять решение использовать стандарты в качестве правил или ссылаться на них в законодательстве.
В чем заключается основная значимость стандартов ИСО для пищевой промышленности?
Стандарты ИСО обеспечивают платформу для разработки практических инструментов через взаимопонимание и сотрудничество со всеми заинтересованными сторонами – от сельскохозяйственных производителей до производителей продуктов питания, лабораторий, регулирующих органов, потребителей и т.
В чем преимущества применения международных стандартов ИСО?
Международные стандарты разрабатываются на основе консенсуса, что положительно влияет на сокращение барьеров в торговле.
Как и кем разрабатываются стандарты ИСО?
Специалисты во всем мире разрабатывают стандарты необходимые для их отрасли. Это значит, что эти стандарты отображают международный опыт и знания
Как можно принять участие в разработке стандартов ИСО?
Стандарты разрабатываются группами специалистов в рамках технического комитета. Эти специалисты назначаются национальными органами-членами ИСО. Для участия в разработке стандарта следует обратиться к своему
национальному представителю
.
Проводит ли ИСО оценку соответствия и сертификацию?
Нет, сама ИСО не проводит оценку соответствия. ИСО разрабатывает стандарты, на соответствие которым проводится сертификация, но некоторые члены ИСО под мандатом своего государства либо коммерческой организации могут осуществлять данный вид деятельности.
ИСО разрабатывает стандарты, на соответствие которым проводится сертификация, но некоторые члены ИСО под мандатом своего государства либо коммерческой организации могут осуществлять данный вид деятельности.
Можно ли использовать логотип ИСО после прохождения сертификации?
Логотип «ISO» является зарегистрированным торговым знаком. Использование этого логотипа запрещено, если нет разрешения правообладателя. ИСО не проводит сертификации, в виду этого не предоставляет логотипов для рекламы, но логотип может быть предоставлен сертифицирующим органом. Подробная информация о правилах использования логотипа представлена по
ссылке.
В каких областях разрабатываются стандарты ИСО?
ИСО с момента создания опубликовала более 22000 международных стандартов, которые распространяются почти на все аспекты технологии и бизнеса. Около 1 000 стандартов разработаны на продукты питания, и связаны с такими темами, как сельскохозяйственная техника, логистика, перевозка, изготовление, маркировка, упаковка и хранение.
Международные стандарты ИСО разработаны в следующих направлениях:
- Стандарты управления качеством
- Стандарты экологического менеджмента
- Стандарты охраны труда
- Стандарты управления энергопотреблением
- Стандарты безопасности пищевых продуктов
- Стандарты IT-безопасности
Кто является членами ИСО?
Членами ИСО являются национальные органы по стандартизации, которые представляют интересы своей страны в ИСО, а также представляют ИСО в своей стране. В РФ таким органом является Федеральное агентство по техническому регулированию и метрологии.
Существует ли официальный сайт ИСО?
Да. Международная организация по стандартизации ИСО – https://www.iso.org/standards.html
Как внедрить систему менеджмента на основе международного стандарта серии ИСО?
Внедрение системы менеджмента на основе международного стандарта серии ИСО можно осуществить своими силами или с помощью консалтинговой компании, где работают профессионалы с многолетним опытом работы.
ссылке
.
Все, что нужно знать об ISO 45001
Независимо от того, являетесь ли Вы сотрудником, менеджером или владельцем бизнеса, Вы стремитесь к достижению общей цели и не хотите, чтобы кто-либо пострадал на работе. Повышение производительности связано с тем, что люди работают на рабочих местах, обеспечивая транспарентность и укрепляя доверие на протяжении всего срока службы. Кроме того, повышение уровня ответственности важно для имиджа компании и репутации.
ISO 45001 является новым стандартом по охране здоровья и безопасности труда на рабочем месте. Данный стандарт стал одним из самых ожидаемых в мире и может способствовать значительному повышению уровня безопасности на рабочем месте.
Данный стандарт стал одним из самых ожидаемых в мире и может способствовать значительному повышению уровня безопасности на рабочем месте.
С учетом того, что ISO 45001 станет частью деловой нормы вне зависимости от того, одобрен ли данный стандарт организацией или нет, важно, чтобы компании были в курсе последних событий. Представители ISOfocus расспросили Кристиана Глезеля, организатора рабочей группы, занимающегося разработкой нового стандарта, и Чарльза Корри, секретаря ИСО/ПК 283, об особенностях нового стандарта.
ISOfocus: Что такое ISO 45001?Фото: Ч. Корри
Ч. Корри, секретарь ИСО/ПК 283, Системы менеджмента охраны здоровья и труда.Фото:К. Глезель
К. Глезель, председатель ИСО/ПК 283 рабочей группы, занимающейся разработкой ISO 45001.К. Глезель и Ч. Корри: ISO 45001 был разработан в переломный момент. В качестве первого в мире международного стандарта, затрагивающего вопросы охраны здоровья и безопасности труда
 Требования с руководством по применению, содержит единую основу для всех организаций, намеренных улучшить свои характеристики. Разработанный для применения высшим руководством, он направлен на обеспечение безопасности на рабочем месте для сотрудников и посетителей. В процессе достижения данных целей крайне важно контролировать все факторы, которые могут спровоцировать заболевание, появление травм и в экстремальных случаях смерть. Посредством смягчения негативного воздействия на физическое, психическое и когнитивное состояние человека ISO 45001 охватывает все данные аспекты.
Требования с руководством по применению, содержит единую основу для всех организаций, намеренных улучшить свои характеристики. Разработанный для применения высшим руководством, он направлен на обеспечение безопасности на рабочем месте для сотрудников и посетителей. В процессе достижения данных целей крайне важно контролировать все факторы, которые могут спровоцировать заболевание, появление травм и в экстремальных случаях смерть. Посредством смягчения негативного воздействия на физическое, психическое и когнитивное состояние человека ISO 45001 охватывает все данные аспекты.Несмотря на то, что ISO 45001 основан на OHSAS 18001, прежнем эталонном показателе охраны здоровья и безопасности труда, это новый стандарт, а не пересмотренный или обновленный, и он должен постепенно дополняться в течение следующих трех лет. Поэтому организациям необходимо пересмотреть текущие методы работы.
Какие основные различия между OHSAS 18001 и ISO 45001?
Существует много различий, но основные изменения заключаются в том, что ISO 45001 концентрируется на взаимодействии между организациями и бизнес-средой, в то время как OHSAS 18001 фокусируется на предотвращении возможных угроз для здоровья и других внутренних проблемах. Но различия существуют и в других аспектах:
Но различия существуют и в других аспектах:
- ISO 45001 основан на процессах – OHSAS 18001 основан на процедурах;
- ISO 45001 динамичен во всех аспектах – OHSAS 18001 не динамичен;
- ISO 45001 рассматривает как риски, так и возможности – OHSAS 18001 рассматривает исключительно риски;
- ISO 45001 включает мнения заинтересованных сторон – OHSAS 18001 не включает.
Данные аспекты способствуют значительным изменениям в восприятии управления безопасностью и охраной труда.
Я сертифицирован по OHSAS 18001. Как мне начать процесс перехода на новый стандарт?
При переходе с OHSAS 18001 необходимо предпринять несколько шагов, прежде чем будет внедрена новая система менеджмента. Если Вы будете следовать нижеприведенной хронологии, то сможете успешно адаптироваться:
Если Вы будете следовать нижеприведенной хронологии, то сможете успешно адаптироваться:
- Проведите анализ заинтересованных сторон (т.е. лиц или организаций, которые могут повлиять на деятельность Вашей организации), а также внутренних и внешних факторов, которые могут отразиться на деятельности организации, и узнайте, как эти риски могут контролироваться посредством Вашей системы управления.
- Определите область деятельности системы, при рассмотрении вопроса о том, каких показателей система менеджмента должна достичь.
- Используйте данную информацию для определения процессов, анализа рисков/ оценки и, что наиболее важно, формирования ключевых показателей (KPI).
После того как Вы адаптируете все данные к инструментам OHSAS 18001, Вы сможете повторно использовать большую часть информации применительно, к Вашей новой системе управления. Таким образом, несмотря на различия в подходах, базовые инструменты будут схожими.
Что мне необходимо знать, если я работаю с ISO 45001 впервые?
Ответ зависит от того, насколько Вы осведомлены о системах менеджмента ИСО. ISO 45001 утверждает Приложение SL, таким образом разделяя структуру высокого уровня (HLS), идентичный основной текст, а также термины и определения с другими недавно пересмотренными стандартами системы управления ИСО, такими как ISO 9001:2015 (менеджмент качества) и ISO 14001:2015 (менеджмент окружающей среды). Если Вы уже знакомы с общей структурой, то основная часть ISO 45001 покажется Вам знакомой, необходимо будет лишь заполнить «пробелы» в системе.
ISO 45001 утверждает Приложение SL, таким образом разделяя структуру высокого уровня (HLS), идентичный основной текст, а также термины и определения с другими недавно пересмотренными стандартами системы управления ИСО, такими как ISO 9001:2015 (менеджмент качества) и ISO 14001:2015 (менеджмент окружающей среды). Если Вы уже знакомы с общей структурой, то основная часть ISO 45001 покажется Вам знакомой, необходимо будет лишь заполнить «пробелы» в системе.
Если это не так, то все может пройти гораздо сложнее. Стандарт достаточно сложен к восприятию, если будет прочитан как обычная книга. Вы должны понимать взаимосвязь между конкретными пунктами. Мой лучший совет – найти хороший учебный курс, который поможет вам раскрыть весь потенциал стандарта. Вы также можете обратиться за консультацией, чтобы получить квалифицированную помощь.
Я сертифицирован как по ISO 9001, так и по ISO 14001. Как ISO 45001 взаимодействует с другими системами менеджмента?
Общая структура ИСО (вышеупомянутая HLS) была целенаправленно разработана для упрощения процесса интеграции новых тем, относящихся к управлению, в существующие системы менеджмента организации. Например, ISO 45001 разработан на основе ISO 14001, поскольку мы знаем, что многие организации объединяют свои функции по охране здоровья и безопасности труда, а также экологические функции.
Например, ISO 45001 разработан на основе ISO 14001, поскольку мы знаем, что многие организации объединяют свои функции по охране здоровья и безопасности труда, а также экологические функции.
Как будет использован ISO 45001?
Мы предвидим, что большинство организаций будут использовать ISO 45001 для построения более эффективной системы управления охраной здоровья и безопасностью труда и только некоторые из них захотят получить более широкое признание, которое можно получить после прохождения сертификации. Не существует обязательных требований для сертификации по стандартам на системы менеджмента ИСО. Наличие официальной системы управления предоставит дополнительные преимущества благодаря внедрению лучших практик. Сертификация является дополнительным инструментом, который продемонстрирует внешним сторонам, что Вы достигли полного соответствия определенному стандарту.
При грамотном внедрении преимущества от ISO 45001 будут безграничными. Помимо того, что стандарт требует, чтобы риски по охране здоровья и безопасности труда были выявлены и проанализированы, в нем также применяется риск-ориентированный подход к самой системе управления охраной труда в целях обеспечения эффективности и постоянного совершенствования согласно постоянно меняющемуся «контексту» организации.
Значки на одежде для стирки: расшифровка
Ярлыки на одежде используют для маркировки текстильных изделий. Нанесенные символы помогают определить правила обработки вещей, их эксплуатации покупателем, предупреждая преждевременный износ и порчу. Ярлык или бирка имеет вид небольшого тканевого лоскута, прикрепленного с изнаночной стороны изделия. На производстве его вшивают в боковой или задний шов, на рубашке ярлык можно найти на воротнике.
Сегодня нанесенные на бирки изображения устанавливаются стандартом международного уровня «ISO 3758-2012. Изделия текстильные. Маркировка по уходу с использованием символов». Существует также уже неактуальный, но применяемый на территории нашей страны — ГОСТ ISO 3758-2014/Изделия текстильные. Маркировка символами по уходу. /ISO 3758-2014. Textiles — Care labelling code using symbols (IDT) », пришедший на смену, ГОСТ ISO 3758-2012.
С начала 2021 года «маркировка» стала носить для легкой промышленности двойной смысл. Каждое изделие должно иметь этикетку с соответствующей информацией о товаре, однако, теперь некоторые разновидности предметов одежды должны иметь и обязательную маркировку для отслеживания единицы товара системой «Честный Знак» (статью про данную маркировку мы писали ранее: «Маркировка текстиля. Кому нужно и зачем?»).
Каждое изделие должно иметь этикетку с соответствующей информацией о товаре, однако, теперь некоторые разновидности предметов одежды должны иметь и обязательную маркировку для отслеживания единицы товара системой «Честный Знак» (статью про данную маркировку мы писали ранее: «Маркировка текстиля. Кому нужно и зачем?»).
Знаки стирки: расшифровка
Сразу определим, что означают знаки стирки – базовые обозначения в маркировке, которые имеют вид таза с водой. Кроме тазика, рисунок включает число (температуру), точки, горизонтальные полосы. Значение знаков для стирки расшифровать просто.
|
Основные символы |
|||
|
|
|
|
|
Число
Число на картинке – это самая максимальная температура воды, в которой можно стирать вещь. Обозначается она в градусах Цельсия, например, 60 °С.
Обозначается она в градусах Цельсия, например, 60 °С.
Одна или две горизонтальные черты
Одинарное, двойное подчеркивание советует пользователю выбрать укороченный режим стирки, а также сократить объем загрузки стиральной машины и интенсивность машинного отжима.
|
Символ |
Условия процесса стирки |
|
|
— максимальная температура стирки 95°С — обычный режим |
|
|
— максимальная температура стирки 95°С — мягкий режим |
|
|
— максимальная температура стирки 60°C — обычный режим |
|
|
— максимальная температура стирки 60°C — мягкий процесс |
|
|
— максимальная температура стирки 50°C — обычный режим |
|
|
— максимальная температура стирки 50°C — мягкий режим |
|
|
— максимальная температура стирки 40°C — обычный режим |
|
|
— максимальная температура стирки 40°C — мягкий режим |
|
|
— максимальная температура стирки 40°C — очень мягкий режим |
|
|
— максимальная температура стирки 30°C — обычный режим |
|
|
— максимальная температура стирки 30°C — мягкий режим |
|
|
— максимальная температура стирки 30°C — очень мягкий режим |
|
|
— ручная стирка — максимальная температура 40°C |
|
|
— стирка запрещена |
Повышать рекомендованный производителем температурный режим стирки категорически запрещено. Если белье не очень грязное, стирать в воде меньшей температуры можно. Так можно не только продлить срок службы одежды, но и сэкономить электроэнергию.
Если белье не очень грязное, стирать в воде меньшей температуры можно. Так можно не только продлить срок службы одежды, но и сэкономить электроэнергию.
Отбеливание
Поскольку речь идет об агрессивном процессе обработки, очень многие изделия имеют бирку с обозначением в виде треугольника. Нередко этот символ перечеркнут – это значит, что отбеливать вещь нельзя.
Что означают другие обозначения с треугольником, смотрите в таблице ниже.
|
Символ |
Процесс отбеливания |
|
|
— разрешено отбеливание любым окисляющим агентом |
|
|
— разрешено отбеливание только кислородсодержащим/нехлорным агентом |
|
|
— не отбеливать |
Прежде чем отбеливать вещь (даже если этикетка разрешает), воспользуйтесь запасным лоскутом ткани. Протестируйте свой отбеливатель на этом кусочке и посмотрите, как поведет себя ткань.
Протестируйте свой отбеливатель на этом кусочке и посмотрите, как поведет себя ткань.
Профессиональная чистка (сухая или влажная)
Химчистка на ярлыке одежды обозначается вариативными символами и их различным сочетанием – квадрат, круг, точки, горизонтальные линии. Разберем, что именно может содержать этикетка.
Буквы
Значок буквы в окружности, говорит о том, что можно использовать растворитель определенного типа.
Одна горизонтальная черта
Подчеркнутое изображение – деликатный режим обработки.
Две горизонтальных черты
Пара линий под кругом – максимально бережная чистка.
Химчистка (сухая профессиональная чистка)
Если изделие можно подвергать сухой обработке, на этикетку наносится круг.
|
Символ |
Режим сухой чистки |
|
— профессиональная сухая чистка в тетрахлорэтилене и во всех растворителях, внесенных в список для символа F — обычный режим |
|
|
— профессиональная сухая чистка в тетрахлорэтилене и во всех растворителях, внесенных в список для символа F — мягкий режим |
|
|
— профессиональная сухая чистка в углеводородах (температура перегонки от 150°C до 210°C, температура воспламенения от 38°C до 70°C) — обычный режим |
|
|
— профессиональная сухая чистка в углеводородах (температура перегонки от 150°C до 210°C, температура воспламенения от 38°C до 70°C) — мягкий режим |
|
| — сухая чистка запрещена |
Аквачистка (мокрая профессиональная чистка)
Изделия, которые можно подвергать мокрой профессиональной чистке, носят букву W, обведенную в окружность.
|
— профессиональная мокрая чистка
— обычный режим |
|
|
— профессиональная мокрая чистка
— мягкий режим |
|
|
— профессиональная мокрая чистка
— очень мягкий режим |
|
| — профессиональная мокрая чистка запрещена |
Если на этикетке вы заметили символ химчистки (круг), подумайте, готовы ли вы регулярно тратить деньги, чтобы правильно ухаживать за вещью. Такие изделия нельзя стирать самостоятельно.
Сушка
Традиционно информацию об отжиме и сушке белья помещают в один значок. Если вещь требует детального ухода, ярлык имеет несколько обозначений.
Сушка в стиральной машине барабанного типа
Горизонтальная черта внутри квадрата – сушить можно на горизонтальной поверхности, вертикальная черта – сушка без отжима.
|
Символ |
Процесс барабанной сушки |
|
|
— барабанная сушка возможна — обычная температура; максимальная температура на выходе 80°C |
|
— барабанная сушка возможна — более низкая температура; максимальная температура на выходе 60°C |
|
| — не применять барабанную сушку |
Естественная сушка
Квадрат без круга – сушка только в естественных условиях.
Обычная сушка
Квадрат без дополнительных обозначений – ограничений в естественной сушке нет.
|
Символ |
Условия естественной сушки |
|
|
— сушка на веревке или вешалке после стирки с отжимом |
|
|
— сушка на веревке или вешалке после стирки без отжима |
|
|
— сушка на плоскости после стирки с отжимом |
|
|
— сушка на плоскости после стирки без отжима |
Сушка в тени (без попадания прямых солнечных лучей)
Об ограничении скажут две косые полоски в углу квадрата.
|
Символ |
Условия естественной сушки |
|
|
— сушка на веревке или вешалке в тени после стирки с отжимом |
|
|
— сушка на веревке или вешалке в тени после стирки без отжима |
|
|
— сушка на плоскости в тени после стирки с отжимом |
|
|
— сушка на плоскости в тени после стирки без отжима |
Символы больше не используются
В таблице ниже приведем символы в теме «сушка», которые больше не применяются.
Если вам мешает этикетка на одежде, можете аккуратно срезать ее, но не выбрасывайте. Сохранив бирку, вы в любое время сможете подсмотреть, можно ли сушить изделие в машинке, можно ли выжимать и выкручивать.
Глажка
Иконки такого типа содержат изображение утюга, показатели температуры, которым соответствуют точки. Подробнее смотрите в таблице ниже.
| Символ | Процесс глажения |
| — гладить при максимальной температуре не более 200°C | |
| — гладить при максимальной температуре не более 150°С | |
|
— гладить при максимальной температуре не более 110°C, без пара — глажение с паром может вызывать необратимые повреждения |
|
Надписи
Чаще всего производители «рассказывают» покупателям об особенностях ухода за изделиями с помощью значков. Однако некоторые бирки на одежде и прочих текстильных изделиях содержат надписи на английском языке, которые имеют следующее значение:
Однако некоторые бирки на одежде и прочих текстильных изделиях содержат надписи на английском языке, которые имеют следующее значение:
- Machine wash – можно стирать в стиральной машине
- Hand wash only – допустима только ручная стирка
- Hot/Cold/Warm wash – стирать в горячей/холодной/теплой воде
- Wash separately – стирать только изолированно от других вещей
- No wash – стирка запрещена
Редко на одежде можно встретить бирку с надписью «Wash with similar colors», что означает «стирать только с вещами похожих цветов». Или, например, «Keep away from fire» — «держать подальше от источников огня».
Ткани
Состав ткани на ярлыках определяет продолжительность срока службы изделия, его внешний вид. Поскольку вещи могут быть изготовлены из одного материала или нескольких, производителю необходимо указывать процентное соотношение компонентов на значках на футболках, платьях, куртках, головных уборах и пр. Такая одежда имеет вшивные бирки или составники.
Такая одежда имеет вшивные бирки или составники.
В числе наиболее популярных волокон:
- хлопок (Co) – натуральное полотно, применяется для пошива легких летних вещей;
- лен (Li) – растительное волокно, гигроскопичное и долговечное.
- нейлон (NY) – синтетическая нить, которая позволяет вещам быстро сохнуть и не мяться;
- полиэстер (PL) – синтетическая нить, которая часто используется тандеме с шерстью и вискозой.
Специфические символы ухода за одеждой в других странах
Выше шла речь о международных обозначениях на одежде, единых значках маркировки, оформление и значение которых не зависит от страны. Однако в стандартах разных государств ранее существовали или все еще существуют особенности, различия, о которых стоит сказать несколько слов.
Австралия
Этикетки изделий австрийских производителей содержат те же «пиктограммы», «квадраты» и «круги». Предупреждающие знаки выделены красным. Другие – синим. Порядок обозначений аналогичен международному.
Предупреждающие знаки выделены красным. Другие – синим. Порядок обозначений аналогичен международному.
Канада
До обновления канадской системы маркировки она включала всего 5 классических символов, которые наносились на бирки в красном (запрещающем), зеленом (разрешающем) и янтарной (предупреждающем) цвете. В 2003 году система была обновлена и подведена к международным стандартам.
Япония
Символы на бирке японского производителя могут быть черными, темно-синими, а символы запрета – только красными на белом фоне. На этикетке изделий, которые обычно не гладят, могут вовсе не нарисовать «утюжок». Исключение составляет лишь значок «глажка запрещена».
Китай
Обозначения на этикетках китайских текстильных изделий практически ничем не отличаются от описанных выше. Одни однотонные, типичные по виду. Однако могут иметь пояснения в виде иероглифов снизу от рисунка.
Европа
Фирменная этикетка одежды из стран Европы может состоять из четырех или пяти символов. Их последовательность строго определена: Стирка, Отбеливание, Глажка, Химчистка, Сушка.
Их последовательность строго определена: Стирка, Отбеливание, Глажка, Химчистка, Сушка.
США
До 1996 года этикетки, над которыми работали американские производители, могли содержать как значки, так и слова, которые являлись инструкцией по уходу за изделием. Современная система Соединенных Штатов Америки содержит только символы, никаких слов, но размещаются они на бирке в знакомом, но строгом порядке: Стирка, Отбеливание, Сушка, Глажка, Предупреждения.
Игнорирование правил ухода за вещью чревато ее преждевременной порчей. Очевидно, можно купить новый пуховик или майку, но гораздо выгоднее не просто знать, что обозначают значки на одежде, но и не забывать подсматривать на этикетку перед тем, как стирать, отбеливать, сушить или отправлять вещь в химчистку.
Расшифровка УЗИ щитовидной железы: норма – МЕДСИ
20.01.2018
Что такое УЗИ?
УЗИ (ультразвуковое исследование, сонография) – процедура для обследования организма посредством ультразвуковых волн. Их частота порядка 20000 Гц, она выше той, которую может воспринять человеческое ухо. Этот анализ безопасен для организма и при необходимости может проводиться часто.
Их частота порядка 20000 Гц, она выше той, которую может воспринять человеческое ухо. Этот анализ безопасен для организма и при необходимости может проводиться часто.
Щитовидная железа вырабатывает йодсодержащие гормоны и хранит йод. Она состоит из двух долей и перешейка и расположена в передней части шеи. Ее правильная работа чрезвычайно важна для нормального функционирования всего организма, а нарушения ведут к ряду заболеваний: рак, базедова болезнь, кретинизм, аденома, микседема и пр.
УЗИ этой железы позволяет узнать, в каком состоянии она находится, есть ли какие-либо патологии или новообразования. Оно показывает изменение ее структуры, из-за которых могут возникнуть различные проблемы со здоровьем.
Как проводится УЗИ?
Перед исследованием не требуется длительной подготовки, но существует несколько правил:
- Рекомендуется не есть за несколько часов до него
- Если предполагается делать допплерографию, необходимо за 3-4 часа принять йодсодержащий препарат
- Перед тем, как лечь на кушетку, нужно снять с шеи все украшения и освободиться от воротника, шарфа и любого другого элемента одежды или декора
Допплер-УЗИ – это вид анализа, который позволяет совместить черно-белое изображение щитовидной железы с цветным отображением кровяного тока. Оно позволяет определить:
Оно позволяет определить:
- Проходимость кровеносных сосудов
- Нарушения их стенок (утончение/утолщение)
- Направление и скорость
- Индекс сопротивления
Сама процедура проходит так:
- Пациент ложится на кушетку
- На его шею наносится специальный крем
- Сонолог водит прибором по области щитовидной железы, и в это время данные передаются на экран и записываются на жесткий диск компьютера
Общее время исследования – порядка 15 минут.
Показания к анализу для взрослого пациента
Ультразвуковое исследование щитовидной железы назначают в таких случаях:
- Пациент бледен и плохо себя чувствует
- Он испытывает боль в горле/шее и при этом не болеет простудными заболеваниями
- Анализы на гормоны показывают нарушения
- Аритмия
- Сонливость, апатия и вялость
- Внезапное ожирение или истощение
- Слишком частые перепады настроения
При планировании беременности также следует провести такое исследование.
Когда УЗИ щитовидной железы необходимо ребенку?
У детей также могут возникнуть проблемы с работой этого органа. Поэтому врач может назначить такое исследование при следующих признаках:
- Во время осмотра у ребенка обнаружено уплотнение на шее
- У него затруднено дыхание, возникает одышка
- Внезапное увеличение/снижение веса
- Аритмия
- Если пациент перенес тяжелое заболевание, при котором возможны осложнения
Существует несколько предпосылок, при наличии которых рекомендуется делать ребенку профилактический осмотр щитовидной железы:
- Проживание на территории с низким содержанием йода в продуктах
- В больших промышленных городах
- Если у ребенка имеется генетическая предрасположенность к проблемам с этим органом
Нормы щитовидной железы
В результате исследования этого органа важным является состояние его долей. Перешеек между ними у здорового человека может как иметься, так и отсутствовать. Главными показателями являются:
Перешеек между ними у здорового человека может как иметься, так и отсутствовать. Главными показателями являются:
- Размеры
- Структура тканей
- Эхогенность
- Наличие или отсутствие новообразований
Размеры в норме
На результатах УЗИ щитовидной железы размеры и объемы имеют важное значение. Максимальный объем должен рассчитывается в соответствии с весом пациента и может составлять от 12,3 см3 при массе до 40 кг до 35 см3 при 110 кг. Часто у женщин он меньше, чем у мужчин, из-за различий некоторых процессов в работе эндокринной системы. Если железа работает правильно, но при этом ее объем на 1 и более см3 больше, то это также считается нормой.
Отдельно рассматриваются параметры долей. На УЗИ щитовидной железы их нормальные размеры должны соответствовать таким категориям:
- Ширина 1,5-2 см
- Длина 2,5-6 см
- Толщина 1-1,5 см
Перешеек может быть от 4 до 8 мм. Размер паращитовидных желез – 2-8 мм. В норме правая доля может быть немного больше левой (изредка – левая больше правой).
Размер паращитовидных желез – 2-8 мм. В норме правая доля может быть немного больше левой (изредка – левая больше правой).
В период беременности размер этого органа может увеличиться, сохраняя нормальное функционирование. Через 3-4 месяца после родов он возвращается в привычное состояние.
У детей до 16-18 щитовидная железа растет постепенно с момента рождения. Может быть увеличена в период полового созревания.
Параметры тканей железы
Структура должна быть зернистой, однородной, железистой и состоять из маленьких фолликул, в которых образуются гормоны. Всего их порядка 30 миллионов.
Ее неоднородность может быть признаком воспалительных заболеваний (диффузный токсический зоб и иные), хотя встречается и у здоровых пожилых людей. Это происходит из-за высокой выработки антител к некоторым ферментам.
Эхогенность
Это параметр, который показывает, как сильно ткани отражают или не отражают ультразвук. Он характеризуется плотностью вещества.
Существует 4 типа эхогенности:
- Анаэхогенный – на мониторе ткани черные, поглощает ультразвук (кровеносные сосуды, доброкачественные образования)
- Изоэхогенный – отражает частично, светло-серый на экране (здоровые ткани)
- Гипоэхогенный – мало отражает, темная область (кисты)
- Гиперэхогенный – полностью отражает, очень светлые части (соединительная ткань)
Если на УЗИ щитовидной железы эхогенность повышена, это может быть признаком иммунных заболеваний.
Новообразования
Анализ помогает выявить различные виды новообразований. Это могут быть:
- Доброкачественные коллоидные узлы, аденомы
- Кисты (содержат жидкость)
- Раковая опухоль
Когда на УЗИ щитовидной железы эхогенность понижена, вероятность наличия кисты или злокачественного образования увеличивается. О нем свидетельствуют размытые границы пятна на сонограмме.
В случае обнаружения таких уплотнений проводят и другие анализы, чтобы уточнить диагноз.
Какие заболевания помогает выявить УЗИ?
В процессе ультразвуковой диагностики врач может обнаружить ряд нарушений. Они определяются в зависимости от размера, объема, структуры тканей и эхогенности. Изменение этих параметров может быть признаком таких заболеваний, как:
- Гипотериоз – уменьшение органа
- Узловой зоб – появление одного или нескольких очагов плотных тканей
- Дифффузно-токсический зоб – чрезмерное увеличение железы
- Воспаление – появление отека и, в некоторых случаях, гноя
- Аденома – доброкачественное опухолевое образование
- Киста – полость, наполненная жидкостью
- Раковая опухоль
Аденому от рака можно отличить по четкости обозначения границ опухоли:
- у доброкачественной они ясно определены и хорошо видны на сонограмме
- у злокачественной – расплывчатые, проросшие в здоровую область
Для установки точного диагноза в таких случаях используют дополнительные методы: анализ крови, КТ, МРТ и другие.
Клиники МЕДСИ предлагают:
- 30 видов ультразвуковых исследований
- современные УЗИ-аппараты экспертного класса – Pro Focus 2202, Philips iU22
- оборудование, которое подходит для обследования детей
- врачи ультразвуковой диагностики и эндокринологи высшей квалификационной категории, кандидаты и доктора медицинских наук
- срочная диагностика
- шаговая доступность – более 20 медицинских центров по всей Москве
Не затягивайте с лечением, обратитесь к врачу прямо сейчас:
- УЗИ
- Прием врача-эндокринолога
Чем же опасна ГЭРБ? Medical On Group Самара
ГЭРБ — гастроэзофагеальная рефлюксная болезнь по праву считается заболеванием 21 века. Она развивается вследствии заброса (рефлюкса) в пищевод из желудка соляной кислоты и/или дуоденального содержимого (желчи и секрета поджелудочной железы). ГЭРБ не только медицинская, но и социальная проблема. У большинства пациентов диагноз остается неверифицированным (неустановленным) длительное время, что приводит к утяжелению ее течения и более позднему началу лечения, а в ряде случаев – возникновению осложнений. Залогом успешного лечения является активное выявление пациентов.
ГЭРБ не только медицинская, но и социальная проблема. У большинства пациентов диагноз остается неверифицированным (неустановленным) длительное время, что приводит к утяжелению ее течения и более позднему началу лечения, а в ряде случаев – возникновению осложнений. Залогом успешного лечения является активное выявление пациентов.
Факторы риска развития ГЭРБ:
- Избыточная масса тела.
- Заболевания желудка с повышенной секреторной функцией.
- Заболевания пищеварительной системы, проявляющиеся дискинетическими (моторными, двигательными) нарушениями.
- Тяжелый физический труд.
- Беременность.
- Курение.
- Заболевания бронхо-легочной системы, проявляющиеся длительным кашлем, приемом теофиллина.
- Системные заболевания, протекающие с поражением соединительной ткани (системная склеродермия, синдром Шегрена, мезенхимопатии и др.).
-
Прием нитропрепаратов, бета-адреноблокаторов, седативных препаратов при заболеваниях сердечно-сосудистой системы.


Признаки ГЭРБ:
- Частая изжога (жжение, которое возникает в желудке или нижней части грудной клетки и поднимается вверх). При физическом напряжении, изменении положения туловища (наклонах, в положении лежа, после еды), при погрешностях в диете (приеме алкоголя, газированных напитков) типичным является усиление изжоги.
- Ощущение кома в горле при глотании, боль в нижней челюсти.
- Отрыжка.
- Боли в эпигастральной области (проекции мечевидного отростка, «под ложечкой») вскоре после еды.
- Срыгивание кислоты.
- Ощущение затруднения или препятствия при прохождении пищи по пищеводу (встречается редко).
Атипичные (внепищеводные) симптомы ГЭРБ:
- Кашель (хронический).
- Ларингит.
-
Эрозия зубной эмали и повышенная чувствительность зубов, кариес.
- Синуситы и фарингиты.
- Астма.
- Аспирационная (вследствие заброса соляной кислоты) пневмония.
- Боль в грудной клетке (преимущественно за грудиной), нарушение ритма.
- Неприятный запах изо рта.
- Икота.
- Кривошея и мышечный спазм.

Опасность этого заболевания состоит в том, что, если болезнь не лечить, то через определенное время кислота разрушает слизистую оболочку пищевода. Это приводит к воспалению и появлению поверхностных изъязвлений (эрозий), а в тяжелых случаях – язвенных дефектов пищевода.
Осложнения ГЭРБ:
- Сужение пищевода.
- Пищевод Баретта (предраковые изменения пищевода).
- Кровотечение из язвы или эрозии пищевода.
- Рак пищевода.
Диагностика ГЭРБ:
Внутрипищеводная pH-метрия. Метод дает объективные данные, свидетельствующие о наличии или отсутствии ГЭРБ. Эффективность метода – 85%.
Метод дает объективные данные, свидетельствующие о наличии или отсутствии ГЭРБ. Эффективность метода – 85%.
Фиброгастродуоденоскопия пищевода (ФЭГДС) — это вид эндоскопии, оценивающий внутреннюю поверхность слизистой оболочки пищевода, а также двенадцатиперстной кишки и желудка. Возможно проведение биопсии. Эффективность метода – 60%.
Лечение ГЭРБ
При лечении ГЭРБ в первую очередь нужно изменить свою диету, стиль жизни, а также правильно принимать лекарства.
При нечастой изжоге используют антациды. При более тяжелой форме заболевания назначают ингибиторы протонной помпы, снижающие секрецию соляной кислоты и способствующие заживлению язв и эрозий. В схему терапии включают прокинетики (средства, нормализующие двигательную функцию верхних отделов пищеварительного тракта, повышающие тонус нижнего пищеводного сфинктера, усиливающие перистальтику пищевода).
Правила для больных ГЭРБ:
- Нормализовать массу тела.
-
Исключить алкоголь, курение, кофе, газированные напитки, выпечку, шоколад, мяту, специи.
- Питаться регулярно маленькими порциями не позже трех часов до сна.
- Исключить острую, жирную, очень горячую, холодную пищу.
- Уменьшить нагрузки на брюшной пресс.
- Не работать в наклон после еды.
- Сильно не затягивать ремни и пояса.
- Приподнять изголовье кровати на 10–15 см.
- Ограничить (при возможности прекратить) прием лекарственных препаратов:

1. Ухудшающих моторику желудочно-кишечного тракта и снижающих тонус нижнего пищеводного сфинктера (бета-блокаторы, нитраты, миотропные спазмолитики, антидепрессанты, теофиллины, прогестерон).
2. Повреждающих слизистую оболочку пищевода (аспирин и другие нестероидные противовоспалительные средства, антихолинэргические препараты, тетрациклины, хинидин, антагонисты кальция 1 и 2 поколений).
При проявлении симптомов ГЭРБ необходима консультация врача-гастроэнтеролога для назначения адекватного лечения, что позволит избежать осложнений.
Автор статьи:
- Гомозкова М. А.
Понравился материал? поделись с друзьями!
Школа инструмента » Маркировка и зернистость наждачной бумаги
Фарида
24 января 2018
Маркировка наждачной бумаги указывает на фракцию и концентрацию абразива.
Отечественный ГОСТ 3647-80 определяет количество зерен разного размера на 1 квадратную единицу, но данная классификация считается устаревшей. Сегодня шкурка шлифовальная соответствует международным стандартам ISO, именно это обозначение можно встретить на обратной стороне изделия. Следует отметить, что данные старого ГОСТа и значения международных стандартов перекликаются между собой. Их соответствие можно проследить в таблице.
Следует отметить, что данные старого ГОСТа и значения международных стандартов перекликаются между собой. Их соответствие можно проследить в таблице.
| Назначение | Маркировка по ГОСТ 3647-80 | Маркировка по ISO-6344 | Размер зерна, мкм | |
| Крупнозернистые | ||||
| Очень грубые работы | 80-Н | P22 | 800-1000 | |
| 63-Н | P24 | 630-800 | ||
| 50-Н | P36 | 500-630 | ||
| Грубые работы | ||||
| 40-Н | P40 | 400-500 | ||
| 32-Н | P46 | 315-400 | ||
| 25-Н | P60 | 250-315 | ||
| Первичная шлифовка | 20-Н | P80 | 200-250 | |
| 16-Н | P90 | 160-200 | ||
| 12-Н | P100 | 125-160 | ||
| 10-Н | P120 | 100-125 | ||
| Окончательная шлифовка мягких пород дерева, старой краски под покраску | 8-Н | P150 | 80-100 | |
| 6-Н | P180 | 63-80 | ||
| Мелкозернистые | ||||
| Окончательная шлифовка твердых пород дерева, шлифовка между покрытиями | 5-Н,М63 | P240 | 50-63 | |
| 4-Н,М50 | P280 | 40-50 | ||
| Полировка финальных покрытий, шлифовка между покрасками, мокрая шлифовка | М40\Н-3 | P400 | 28-40 | |
| М28\Н-2 | P600 | 20-28 | ||
| Шлифовка металла, пластиков, керамики, мокрая шлифовка | М20\Н-1 | P1000 | 14-20 | |
| Еще более тонкая шлифовка, полировка | М14 | P1200 | 10-14 | |
| М10/Н-0 | P1500 | 7-10 | ||
| М7\Н-01 | P2000 | 5-7 | ||
| М5\Н-00 | P2500 | 3-5 | ||
Основная разница старого и нового стандартов заключается в разном движении номеров в маркировке:
В ГОСТ 3647-80 значения крупности и плотности зерен уменьшаются, что вполне логично;
В новом стандарте ISO 6344 (ГОСТ 52318-2005) номер изделия увеличивается с уменьшением значения в маркировке.
Расшифровка зернистости
Буква «Р» в обозначении указывает на зернистость. Чем больше значение, стоящее за буквой, тем мельче фракция шкурки.
Р400 – известная нулёвка. Р600…Р2500 имеют мелкие фракции и почти гладкую поверхность, их зернистость не так ощутима наощупь. Такую наждачку используют для финишной полировки в промышленности.
В старом ГОСТе дела обстояли иначе. На примере 10-Н: первая цифра означает, что на поверхность изделия нанесен абразив крупностью, соответствующей ситу с ячейкой 100 мкм. Чем меньше показатель, тем меньше размер просева.
Другие обозначения
Виды наждачной бумаги различаются по составу основы и свойствам зерен. Эту информацию также отражают в маркировке изделия.
Литера «Л» указывает на листовую форму выпуска изделия. Рулонная не обозначается никак.
Буквой «М» обозначается водостойкая наждачная бумага.
Изделие с маркировкой «П» предназначено для шлифовки сухих изделий без контакта с влагой.
«1» – абразив для мягких поверхностей, «2» – для жестких.
Теги: наждачная бумага, маркировка
Секвенирование РНК методом Iso-Seq
Перейти к содержимому
Исследования транскриптома человека
Узнайте, как изменения в использовании изоформ способствуют фенотипическим различиям между здоровьем и болезнью.
Учить больше
Транскриптомные исследования растений и животных
Продвинутые программы селекции и фундаментальные исследования с полноразмерным секвенированием кДНК, не требующим эталонного генома.
Учить больше
Исследования транскриптома отдельных клеток
Визуализируйте биологию с еще более высоким разрешением, захватывая всестороннюю и полную последовательность транскриптов из отдельных клеток.
Учить больше
Введение в секвенирование РНК с использованием метода Iso-Seq
С помощью длинных и точных считываний HiFi можно охарактеризовать все разнообразие транскриптома:
- См. альтернативные начальный и конечный сайты
- Характеристика событий монтажа
- Идентификация аллель-специфических изоформ
- Выражение профиля при разрешении изоформы
- Обнаружение гибридных генов
- Прогнозирование открытых кадров чтения полной длины
- …и более
«Более 50% изоформ, надежно обнаруженных с помощью Iso-Seq, не могут быть легко воспроизведены с использованием стандартной РНК-seq, что подчеркивает преимущество секвенирования с длинным считыванием для характеристики разнообразия изоформ». 1
— Леунг, и др. , (2021)
1. Leung, et al.
 (2021) Полноразмерное секвенирование транскриптов коры головного мозга человека и мыши выявляет широко распространенное изоформное разнообразие и альтернативный сплайсинг. Представитель ячейки . 37(7):110022.
(2021) Полноразмерное секвенирование транскриптов коры головного мозга человека и мыши выявляет широко распространенное изоформное разнообразие и альтернативный сплайсинг. Представитель ячейки . 37(7):110022.Секвенирование РНК с анализом Iso-Seq в действии
Статья
Новый метод фазирования изоформ прослеживает различия между родителями и потомками у кукурузы
Узнайте, как ученые из Колд-Спринг-Харбор стремились обеспечить, чтобы потомство кукурузы могло превзойти своих «родителей».
Читать далее
Масштабируемое секвенирование изоформ РНК с использованием внутримолекулярного мультиплексирования кДНК
Для масштабируемого полноразмерного секвенирования РНК мы разработали мультиплексированные массивы для секвенирования…
Смотреть видео
Энтузиасты анализа Iso-Seq делятся победами в исследованиях на виртуальном мероприятии
Десятки самопровозглашенных фанатов анализа Iso-Seq и других любопытных исследователей собрались…
Читать далее
Ссылка на транскриптом уровня изоформы помогает креветкам предотвратить болезни
Креветки являются одним из самых быстрорастущих продуктов питания в мире, но также и одним из наиболее уязвимых.
Читать далее
УЗНАТЬ БОЛЬШЕ
Знаете ли вы, что у нас есть обширная библиотека статей, отчетов, документов и видео, связанных с секвенированием РНК методом Iso-Seq?
Исследуйте ресурсы
ФОРМИРОВАНИЕ БУДУЩЕГО
Исследователи Университета штата Вашингтон используют анализ Iso-Seq, чтобы пролить свет на тайну метаболизма бурых медведей.
Читать далее
Краткий обзор рабочих процессов приложений Iso-Seq
| Аннотация генома | Весь транскриптом | Целевой Iso-Seq | Одноклеточный транскриптом | |
|---|---|---|---|---|
| Цель | Качественная аннотация генома | Эталонный транскриптом для конкретного образца для анализа экспрессии изоформ с соответствующими короткими данными считывания или без них | Количественная характеристика изоформ выбранных генов | Идентифицировать изоформы, специфичные для типа клеток |
| Подготовка библиотеки | Протокол Iso-Seq | Протокол Iso-Seq | Скоро | Протокол Iso-Seq для одной клетки |
| Секвенирование | 1 SMRT Cell 8M (рекомендуется мультиплексирование до 12 тканей) | 1 ячейка SMRT 8M на образец | 1 ячейка SMRT 8M (рекомендуется мультиплексировать до 24 выборок) | 1 ячейка SMRT 8M (рекомендуется добавить соответствующие короткие данные для чтения) |
| Анализ | Анализ Iso-Seq (SMRTLink) или командная строка (isoseq. how) how) | Анализ Iso-Seq с последующими инструментами сообщества (SQANTI3, tappAS, Kallisto) | Анализ Iso-Seq с последующими инструментами сообщества (SQANTI3, tappAS, Kallisto) | Одноклеточный анализ |
Преимущества секвенирования РНК с помощью метода
Iso-Seq
Метод Iso-Seq, основанный на чтении HiFi, раскрывает биологию, которую упускают другие подходы.
Метод Iso-Seq позволяет получать полноразмерные изоформы транскриптов, сборка не требуется
В отличие от методов РНК-секвенирования с коротким чтением, метод Iso-Seq считывает весь транскрипт, захватывая даже нетранслируемые области.
- Обнаружение альтернативного полиаденилирования
- Обнаружение использования дифференциальных изоформ (DIU)
- Получить информацию о фазах на уровне изоформ
Ван, Б. и др. (2020) Фазирование вариантов и гаплотипическое выражение на основе долговременного секвенирования у кукурузы. Коммун Биол .
Рисунок 3e из Leung et al. (2021)
Данные Iso-Seq являются исчерпывающими
Охарактеризовать разнообразие изоформ всего транскриптома, которое может выявить сложный альтернативный сплайсинг, события слияния и прочтения транскрипции.
Анализ Iso-Seq предлагает беспрецедентную информацию на уровне отдельных клеток
Используйте любую одноклеточную платформу для создания полноразмерной кДНК в качестве исходных данных для SMRT-секвенирования и получите все те же преимущества Iso-Seq для объемной кДНК — полная последовательность транскриптов, фазирование на уровне изоформ и профилирование изоформ — в одноклеточных разрешающая способность.
Джоглекар А, и др. (2021) Область мозга с пространственным разрешением и атлас изоформ, специфичных для типа клеток мозга постнатальной мыши. Нац.коммун.
ИНФОРМАЦИЯ О РАЗРАБОТКЕ ТЕХНОЛОГИЙ
Метод MAS-ISO-Seq обеспечивает более чем 15-кратную пропускную способность за счет конкатенации
Наши технологии постоянно развиваются. Данные MAS-ISO-Seq используют преимущества длинных и точных длин считывания HiFi для увеличения пропускной способности Iso-Seq более чем в 15 раз за счет мультиплексированных массивов молекул кДНК.
Посмотрите выступление MAS-ISO-Seq на ASHG или прочитайте препринт.
Общие вопросы о методе PacBio Iso-Seq и секвенировании РНК
Метод Iso-Seq относится к секвенированию полноразмерной кДНК с использованием технологии секвенирования PacBio SMRT. Полноразмерные кДНК могут быть получены из эукариотической РНК, бактериальной РНК или даже вирусной РНК. Поскольку полноразмерная кДНК секвенируется в одном прочтении HiFi, сборка не требуется.
Метод Iso-Seq и рабочий процесс биоинформатики позволяют получать высококачественные полноразмерные последовательности транскриптов размером 10 КБ или более. Высокая точность считывания HiFi гарантирует возможность идентификации SNP, а также UMI и штрих-кодов, используемых для исследований отдельных клеток.
Рабочий процесс биоинформатического анализа Iso-Seq не требует эталонного генома, хотя, если эталонный геном доступен, его можно использовать для обратного сопоставления полноразмерных транскриптов с геномом.
Метод Iso-Seq использовался для обнаружения слитых генов рака. Примеры включают идентификацию слияния IGH-DUX4 при остром В-клеточном лимфобластном лейкозе и слияния 3-х прыжков в клеточной линии SKBR3.
Метод Iso-Seq для одиночных клеток (scIso-Seq) был применен к различным платформам для одиночных клеток, которые генерируют полноразмерные молекулы из одиночных клеток и обнаруживают специфические для клеточного типа изоформы, не поддающиеся обнаружению с помощью краткой информации считывания на уровне гена. в таких образцах, как постнатальный мозг мыши и стареющий мозг с синдромом Дауна.
в таких образцах, как постнатальный мозг мыши и стареющий мозг с синдромом Дауна.
Было показано, что метод Iso-Seq имеет согласованную экспрессию на уровне генов с соответствующими данными короткого считывания, при этом он способен идентифицировать дифференциальное использование изоформ (DIU), которое невозможно наблюдать с помощью сборок транскриптов на основе коротких чтений. Имея существующие данные RNA-Seq, метод Iso-Seq может предоставить эталонный транскриптом изоформы для конкретного образца, который улучшает количественный анализ изоформ.
СистемыSequel II могут генерировать более длинные считывания с большей точностью и пропускной способностью при значительно меньших затратах. Изучите наши платформы секвенирования для полноразмерного секвенирования кДНК.
Учить больше
Поговорите с экспертом
Если у вас есть вопрос, вам нужно проверить статус заказа или вы заинтересованы в покупке инструмента, мы здесь, чтобы помочь.
Использование PacBio Iso-Seq для открытия новых транскриптов и генов реакций на абиотический стресс у Oryza sativa L.
1. Ламауи М., Джемо М., Датла Р., Беккауи Ф. Тепловые и засушливые стрессы сельскохозяйственных культур и подходы к их смягчение. Фронт. хим. 2018;6:26. doi: 10.3389/fchem.2018.00026. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
2. Zhao C., Liu B., Piao S., Wang X., Lobell D.B., Huang Y., Huang M., Yao Y., Бассу С., Сиаис П. и др. Повышение температуры снижает глобальные урожаи основных сельскохозяйственных культур по четырем независимым оценкам. проц. Натл. акад. науч. США. 2017;114:9326. doi: 10.1073/pnas.1701762114. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
3. Иидзуми Т., Раманкутти Н. Изменения изменчивости урожайности основных культур за 1981–2010 гг., объясняемые изменением климата. Окружающая среда. Рез. лат. 2016;11:034003. doi: 10.1088/1748-9326/11/3/034003. [CrossRef] [Google Scholar]
4. Доусон Т.П., Перриман А.Х., Осборн Т.М. Моделирование воздействия изменения климата на глобальную продовольственную безопасность. Клим. Чанг. 2016; 134:429–440. doi: 10.1007/s10584-014-1277-y. [Перекрестная ссылка] [Академия Google]
Клим. Чанг. 2016; 134:429–440. doi: 10.1007/s10584-014-1277-y. [Перекрестная ссылка] [Академия Google]
5. Пэн С., Хуан Дж., Шихи Дж. Э., Лаза Р. К., Висперас Р. М., Чжун С., Сентено Г. С., Хуш Г. С., Кассман К. Г. Урожайность риса снижается с повышением ночной температуры из-за глобального потепления. проц. Натл. акад. науч. США. 2004; 101:9971–9975. doi: 10.1073/pnas.0403720101. [Статья PMC бесплатно] [PubMed] [CrossRef] [Google Scholar]
6. Stein J.C., Yu Y., Copetti D., Zwickl D.J., Zhang L., Zhang C., Chougule K., Gao D., Iwata A., Goicoechea J.L. и соавт. Геномы 13 одомашненных и диких родственников риса подчеркивают генетическое сохранение, оборот и инновации для всего рода Oryza. Нац. Жене. 2018; 50: 285–296. doi: 10.1038/s41588-018-0040-0. [PubMed] [CrossRef] [Google Scholar]
7. ФАО . Продовольственные системы для лучшего питания. ФАО; Рим, Италия: 2013. [Google Scholar]
8. Проект генома риса Проект 3000 геномов риса. ГигаНаука. 2014;3:7. [Бесплатная статья PMC] [PubMed] [Google Scholar]
9. Махеш Х.Б., Ширке М.Д., Сингх С., Раджамани А., Хитталмани С., Ван Г.Л., Гоуда М. Сборка генома риса Indica , аннотация и анализ генов устойчивости к пирикуляриозу. БМС Геном. 2016;17:242. doi: 10.1186/s12864-016-2523-7. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
Махеш Х.Б., Ширке М.Д., Сингх С., Раджамани А., Хитталмани С., Ван Г.Л., Гоуда М. Сборка генома риса Indica , аннотация и анализ генов устойчивости к пирикуляриозу. БМС Геном. 2016;17:242. doi: 10.1186/s12864-016-2523-7. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
10. Гофф С.А., Рике Д., Лан Т.Х., Престинг Г., Ван Р., Данн М., Глейзбрук Дж., Сешнс А., Оллер П., Варма Х. и др. Проект последовательности генома риса ( Oryza sativa L. ssp. japonica ) Science. 2002; 296: 92–100. doi: 10.1126/science.1068275. [PubMed] [CrossRef] [Google Scholar]
11. Wang W., Mauleon R., Hu Z., Chebotarov D., Tai S., Wu Z., Li M., Zheng T., Fuentes R.R., Zhang Ф. и др. Геномная изменчивость 3010 различных образцов риса, выращиваемого в Азии. Природа. 2018; 557: 43–49. doi: 10.1038/s41586-018-0063-9. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
12. Du H., Yu Y., Ma Y., Gao Q., Cao Y., Chen Z., Ma B., Qi M. , Ли Ю., Чжао С. и др. Секвенирование и сборка de novo почти полного генома риса indica . Нац. коммун. 2017;8:15324. doi: 10.1038/ncomms15324. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
и др. Секвенирование и сборка de novo почти полного генома риса indica . Нац. коммун. 2017;8:15324. doi: 10.1038/ncomms15324. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
13. Zhang J., Chen L.L., Sun S., Kudrna D., Copetti D., Li W., Mu T., Jiao W.B., Xing Ф., Ли С. и др. Корпус два indica эталонные геномы риса с данными долгосрочного считывания PacBio и секвенирования парных концов Illumina. науч. Данные. 2016;3:160076. doi: 10.1038/sdata.2016.76. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
14. Сакаи Х., Канамори Х., Араи-Китисе Ю., Шибата-Хатта М., Эбана К., Ооно Ю., Курита К. , Fujisawa H., Katagiri S., Mukai Y., et al. Конструирование псевдомолекулярных последовательностей сорта риса aus Kasalath для сравнительной геномики азиатского культивируемого риса. Рез. ДНК 2014;21:397–405. doi: 10.1093/dnares/dsu006. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
15. McNally K. L., Childs K.L., Bohnert R., Davidson R.M., Zhao K., Ulat VJ., Zeller G., Clark R.M., Hoen D.R., Bureau Т.Е. и др. Полногеномная изменчивость SNP выявляет взаимосвязь между местными сортами и современными сортами риса. проц. Натл. акад. науч. США. 2009;106:12273–12278. doi: 10.1073/pnas.0
L., Childs K.L., Bohnert R., Davidson R.M., Zhao K., Ulat VJ., Zeller G., Clark R.M., Hoen D.R., Bureau Т.Е. и др. Полногеномная изменчивость SNP выявляет взаимосвязь между местными сортами и современными сортами риса. проц. Натл. акад. науч. США. 2009;106:12273–12278. doi: 10.1073/pnas.0
16. Гаррис А.Дж., Маккауч С.Р., Кресович С. Структура популяции и ее влияние на разнообразие гаплотипов и неравновесие по сцеплению, окружающее
.0062 xa5 местонахождение риса ( Oryza sativa L.) Генетика. 2003; 165: 759–769. [Бесплатная статья PMC] [PubMed] [Google Scholar]
17. Gamuyao R., Chin J.H., Pariasca-Tanaka J., Pesaresi P., Catausan S., Dalid C., Slamet-Loedin I., Tecson-Mendoza EM, Wissuwa M., Heuer S. Протеинкиназа Pstol1 из традиционного риса обеспечивает устойчивость к дефициту фосфора. Природа. 2012; 488:535. doi: 10.1038/nature11346. [PubMed] [CrossRef] [Google Scholar]
18. Xu K., Xu X., Fukao T., Canlas P., Maghirang-Rodriguez R., Heuer S. Sub1A представляет собой ген, подобный фактору реакции на этилен, который придает рису толерантность к затоплению. Природа. 2006; 442: 705–708. doi: 10.1038/nature04920. [PubMed] [CrossRef] [Google Scholar]
Xu K., Xu X., Fukao T., Canlas P., Maghirang-Rodriguez R., Heuer S. Sub1A представляет собой ген, подобный фактору реакции на этилен, который придает рису толерантность к затоплению. Природа. 2006; 442: 705–708. doi: 10.1038/nature04920. [PubMed] [CrossRef] [Google Scholar]
19. Хаттори Ю., Нагаи К., Фурукава С., Сонг Х.Дж., Кавано Р., Сакакибара Х., Ву Дж., Мацумото Т., Йошимура А., Китано Х. и др. Факторы отклика на этилен SNORKEL1 и SNORKEL2 позволяют рису адаптироваться к глубоководью. Природа. 2009; 460:1026–1030. doi: 10.1038/nature08258. [PubMed] [CrossRef] [Академия Google]
20. Baltazar M.D., Ignacio JCI, Thomson MJ, Ismail A.M., Mendioro M.S., Septiningsih E.M. Картирование QTL устойчивости к анаэробному прорастанию от IR64 и местного сорта Nanhi с использованием генотипирования SNP. Эвфитика. 2014; 197: 251–260. doi: 10.1007/s10681-014-1064-x. [CrossRef] [Google Scholar]
21. Baltazar MD, Ignacio JCI, Thomson MJ, Ismail AM, Mendioro M.S., Septiningsih EM. Картирование QTL устойчивости к анаэробному прорастанию риса из IR64 и aus ландрас Харсу 80А. Порода. науч. 2019;69:227–233. doi: 10.1270/jsbbs.18159. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
Картирование QTL устойчивости к анаэробному прорастанию риса из IR64 и aus ландрас Харсу 80А. Порода. науч. 2019;69:227–233. doi: 10.1270/jsbbs.18159. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
22. Bernier J., Kumar A., Venuprasad R., Spaner D., Verulkar S., Mandal N.P., Sinha P.K., Peeraju P., Dongre П.Р., Махто Р.Н. и др. Характеристика влияния QTL на засухоустойчивость риса, qtl12.1, в различных средах на Филиппинах и в восточной Индии. Эвфитика. 2009; 166: 207–217. doi: 10.1007/s10681-008-9826-y. [Перекрестная ссылка] [Академия Google]
23. Slabaugh E., Desai J.S., Sartor R.C., Lawas L.M.F., Jagadish S.V.K., Doherty C.J. На анализ дифференциальной экспрессии генов и альтернативного сплайсинга существенное влияние оказывает выбор эталонного генома. РНК. 2019;25:669–684. doi: 10.1261/РНК.070227.118. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
24. Rhoads A., Au K.F. Секвенирование PacBio и его приложения. Геном. протеом. биоинф. 2015;13:278–289. doi: 10.1016/j.gpb.2015.08.002. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
протеом. биоинф. 2015;13:278–289. doi: 10.1016/j.gpb.2015.08.002. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
25. Абдель-Гани С.Э., Гамильтон М., Якоби Дж.Л., Нгам П., Девитт Н., Шилки Ф., Бен-Гур А., Редди А.С. Обзор транскриптома сорго с использованием длинных чтений одиночных молекул. Нац. коммун. 2016;7:11706. doi: 10.1038/ncomms11706. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
26. Clavijo B.J., Venturini L., Schudoma C., Accinelli G.G., Kaithakottil G., Wright J., Borrill P., Kettleborough G., Heavens Д., Чепмен Х. и др. Улучшенная сборка и аннотация генома аллогексаплоидной пшеницы идентифицируют полные семейства агрономических генов и предоставляют геномные доказательства хромосомных транслокаций. Геном Res. 2017;27:885–896. doi: 10.1101/гр.217117.116. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
27. Dong L., Liu H., Zhang J., Yang S., Kong G., Chu J.S., Chen N., Wang D. Single Секвенирование транскриптов молекул в реальном времени облегчает аннотацию генома мягкой пшеницы и исследование транскриптома зерна. БМС Геном. 2015;16:1039. doi: 10.1186/s12864-015-2257-y. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
БМС Геном. 2015;16:1039. doi: 10.1186/s12864-015-2257-y. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
Обзор сложного транскриптома из высокополиплоидного генома сахарного тростника с использованием полноразмерного секвенирования изоформ и сборки de novo из короткого секвенирования. БМС Геном. 2017;18:395. doi: 10.1186/s12864-017-3757-8. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
29. Feng S., Xu M., Liu F., Cui C., Zhou B. Реконструкция полноразмерного атласа транскриптома с использованием PacBio Iso- Seq дает представление об альтернативном сплайсинге Gossypium australe . BMC Растение Биол. 2019;19:365. doi: 10.1186/s12870-019-1968-7. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
30. Carvalho D.S., Nishimwe A.V., Schnable J.C. Сборка транскриптома IsoSeq травы C3 panicoid предоставляет инструменты для изучения эволюционных изменений в Паникоидные . Завод Директ. 2020;4:e00203. doi: 10.1002/pld3. 203. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
203. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
31. Chao Y., Yuan J., Li S., Jia S., Han L., Xu L. Анализ транскриптов и изоформ сплайсинга выделены красным клевера ( Trifolium pratense L.) с помощью долговременного секвенирования одной молекулы. BMC Растение Биол. 2018;18:300. doi: 10.1186/s12870-018-1534-8. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
32. Алам Р., Хаммель М., Йенг Э., Локк А.М., Игнасио Дж.К.И., Балтазар М.Д., Цзя З., Исмаил А.М., Септинингсих Э.М., Bailey-Serres J. Локусы устойчивости к наводнениям SUBMERGENCE 1 и ANAEROBIC GERMINATION 1 взаимодействуют в проростках, выращенных под водой. Завод Директ. 2020;4:e00240. doi: 10.1002/pld3.240. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
33. Шааршмидт С., Лавас Л.М.Ф., Глаубиц У., Ли Х., Эрбан А., Копка Дж., Джагадиш С.В.К., Хинча Д.К., Цутер Э. Сезон влияет на урожайность и метаболические профили риса ( Oryza sativa ) в условиях высокой ночной температурной нагрузки в полевых условиях. ИЖМС. 2020;21:3187. doi: 10.3390/ijms210
ИЖМС. 2020;21:3187. doi: 10.3390/ijms210
34. Лавас Л.М.Ф., Ши В., Йошимото М., Хасегава Т., Хинча Д.К., Зутер Э., Джагадиш С.В.К. Комбинированное воздействие засухи и теплового стресса в период цветения и налива зерна у контрастных сортов риса, выращенных в полевых условиях. Полевые культуры Res. 2018;229: 66–77. doi: 10.1016/j.fcr.2018.09.009. [CrossRef] [Google Scholar]
35. Li X., Lawas L.M., Malo R., Glaubitz U., Erban A., Mauleon R., Heuer S., Zuther E., Kopka J., Hincha D.K., et al. др. Метаболические и транскриптомные сигнатуры цветочных органов риса показывают, что сахарное голодание является фактором репродуктивной недостаточности в условиях стресса от жары и засухи. Окружающая среда растительной клетки. 2015;38:2171–2192. doi: 10.1111/pce.12545. [PubMed] [CrossRef] [Google Scholar]
36. Glaubitz U., Li X., Köhl K.I., van Dongen J.T., Hincha D.K., Zuther E. Дифференциальные физиологические реакции различных сортов риса ( Oryza sativa ) к повышенной ночной температуре во время вегетативного роста. Функц. биол. растений 2014;41:437. дои: 10.1071/FP13132. [PubMed] [CrossRef] [Google Scholar]
Функц. биол. растений 2014;41:437. дои: 10.1071/FP13132. [PubMed] [CrossRef] [Google Scholar]
37. Лейнонен Р., Сугавара Х., Шамуэй М. Сотрудничество с международной базой данных нуклеотидных последовательностей, Архив чтения последовательности. Нуклеиновые Кислоты Res. 2011;39:D19–D21. doi: 10.1093/nar/gkq1019. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
38. Camacho C., Coulouris G., Avagyan V., Ma N., Papadopoulos J., Bealer K., Madden T.L. BLAST+: Архитектура и приложения. БМК Биоинформ. 2009 г.;10:421. doi: 10.1186/1471-2105-10-421. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
39. Ardui S., Ameur A., Vermeesch J.R., Hestand M.S. Секвенирование отдельных молекул в реальном времени (SMRT) достигло совершеннолетия: приложения и утилиты для медицинской диагностики. Нуклеиновые Кислоты Res. 2018;46:2159–2168. doi: 10.1093/nar/gky066. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
40. Финн Р.Д., Эттвуд Т.К., Бэббит П. К., Бейтман А., Борк П., Бридж А.Дж., Чанг Х.Ю., Достаньи З., Эль-Гебали С. ., Фрейзер М. и соавт. InterPro в 2017 году — помимо аннотаций семейства белков и доменов. Нуклеиновые Кислоты Res. 2017;45:D190–D199. doi: 10.1093/nar/gkw1107. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
К., Бейтман А., Борк П., Бридж А.Дж., Чанг Х.Ю., Достаньи З., Эль-Гебали С. ., Фрейзер М. и соавт. InterPro в 2017 году — помимо аннотаций семейства белков и доменов. Нуклеиновые Кислоты Res. 2017;45:D190–D199. doi: 10.1093/nar/gkw1107. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
41. Schwacke R., Ponce-Soto G.Y., Krause K., Bolger A.M., Arsova B., Hallab A., Gruden K., Stitt M. , Bolger ME, Usadel B. MapMan4: Уточненная структура классификации белков и аннотаций, применимая к анализу мультиомных данных. Мол. Завод. 2019;12:879–892. doi: 10.1016/j.molp.2019.01.003. [PubMed] [CrossRef] [Google Scholar]
42. Verma G., Dhar Y.V., Srivastava D., Kidwai M., Chauhan P.S., Bag S.K., Asif M.H., Chakrabarty D. Полногеномный анализ семейства генов дегидринов риса : Его эволюционная консервативность и характер экспрессии в ответ на дегидратационный стресс, индуцированный ПЭГ. ПЛОС ОДИН. 2017;12:e0176399. doi: 10.1371/journal.pone.0176399. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
43. Graether S.P., Boddington K.F. Расстройство и функция: обзор семейства белков дегидринов. Фронт. Растениевод. 2014;5:576. doi: 10.3389/fpls.2014.00576. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
Graether S.P., Boddington K.F. Расстройство и функция: обзор семейства белков дегидринов. Фронт. Растениевод. 2014;5:576. doi: 10.3389/fpls.2014.00576. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
44. Hundertmark M., Hincha D.K. Белки LEA (поздний эмбриогенез) и кодирующие их гены у Arabidopsis thaliana . БМС Геном. 2008; 9:118. дои: 10.1186/1471-2164-9-118. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
45. Мадейра Ф., Пак Ю.М., Ли Дж., Бусо Н., Гур Т., Мадхусуданан Н., Басуткар П., Тиви А.Р.Н., Поттер С.К., Финн Р.Д. и соавт. API инструментов поиска и анализа последовательности EMBL-EBI в 2019 г. Nucleic Acids Res. 2019;47:W636–W641. doi: 10.1093/nar/gkz268. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
46. Dong X., Gao Y., Chen W., Wang W., Gong L., Liu X. Пространственно-временное распределение феноламидов и генетика естественная вариация гидроксициннамоилспермидина в рисе. Мол. Завод. 2015; 8: 111–121. doi: 10.1016/j.molp.2014.11.003. [PubMed] [CrossRef] [Академия Google]
doi: 10.1016/j.molp.2014.11.003. [PubMed] [CrossRef] [Академия Google]
47. Workman R.E., Myrka A.M., Wong G.W., Tseng E., Welch KC, Jr., Timp W. Полноразмерное секвенирование одной молекулы дает представление об экстремальном метаболизме колибри с красным горлом Archilochus colubris . ГигаНаука. 2018; 7 doi: 10.1093/gigascience/giy009. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
48. Li J., Harata-Lee Y., Denton MD, Feng Q., Rathjen J.R., Qu Z., Adelson D.L. Реконструкция полноразмерного транскриптома из 9 референсных геномов без использования длинного считывания.0062 Astragalus membranaceus выявляет варианты транскриптов, участвующих в биосинтезе биоактивных соединений. Сотовый Дисков. 2017;3:17031. doi: 10.1038/celldisc.2017.31. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
49. Xie L., Teng K., Tan P., Chao Y., Li Y., Guo W., Han L. PacBio одиночная молекула Секвенирование с длительным чтением проливает новый свет на транскрипты и изоформы сплайсинга многолетнего райграса. Мол. Жене. Геном. 2020; 295: 475–489. doi: 10.1007/s00438-019-01635-y. [PubMed] [CrossRef] [Академия Google]
Мол. Жене. Геном. 2020; 295: 475–489. doi: 10.1007/s00438-019-01635-y. [PubMed] [CrossRef] [Академия Google]
50. Куо Р.И., Ченг Ю., Смит Дж., Арчибальд А.Л., Берт Д.В. Освещение темной стороны транскриптома человека с помощью анализа TAMA Iso-Seq. bioRxiv. 2019:780015. doi: 10.1101/780015. [CrossRef] [Google Scholar]
51. Zhang G., Sun M., Wang J., Lei M., Li C., Zhao D., Huang J., Li W., Li S., Li J. , и другие. Полноразмерное секвенирование кДНК PacBio, интегрированное с чтением RNA-seq, значительно улучшает обнаружение транскриптов сплайсинга в рисе. Плант Дж. 2019; 97: 296–305. doi: 10.1111/tpj.14120. [PubMed] [CrossRef] [Академия Google]
52. Ван Б., Регулски М., Ценг Э., Олсон А., Гудвин С., МакКомби В.Р., Уэр Д. Сравнительный транскрипционный ландшафт кукурузы и сорго, полученный с помощью секвенирования одиночных молекул. Геном Res. 2018; 28: 921–932. doi: 10.1101/gr.227462.117. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
53. Ван Б. , Ценг Э., Регулски М., Кларк Т.А., Хон Т., Цзяо Ю., Лу З., Олсон А., Стейн Дж. К., Уэр Д. Раскрытие сложности транскриптома кукурузы с помощью долговременного секвенирования одной молекулы. Нац. коммун. 2016;7:11708. doi: 10.1038/ncomms11708. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
, Ценг Э., Регулски М., Кларк Т.А., Хон Т., Цзяо Ю., Лу З., Олсон А., Стейн Дж. К., Уэр Д. Раскрытие сложности транскриптома кукурузы с помощью долговременного секвенирования одной молекулы. Нац. коммун. 2016;7:11708. doi: 10.1038/ncomms11708. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
54. Куо Р. ТАМА: Аннотация транскриптома с помощью модульных алгоритмов. [(по состоянию на 20 февраля 2019 г.)]; Доступно в Интернете: https://github.com/GenomeRIK/tama
55. Тунг Л.Х., Шао М., Кингсфорд К. Количественная оценка преимуществ, предлагаемых сборкой транскриптов при длинных чтениях с одной молекулой. bioRxiv. 2019: 632703. дои: 10.1101/632703. [Статья бесплатно PMC] [PubMed] [CrossRef] [Google Scholar]
56. Олсен А.Н., Манди Дж., Скривер К. Пептомика, идентификация новых катионных пептидов арабидопсиса с консервативными мотивами последовательности. Силико Биол. 2002; 2: 441–451. [PubMed] [Академия Google]
57. Mundy J., Chua N.H. Абсцизовая кислота и водный стресс индуцируют экспрессию нового гена риса. EMBO J. 1988; 7: 2279–2286. doi: 10.1002/j.1460-2075.1988.tb03070.x. [Статья бесплатно PMC] [PubMed] [CrossRef] [Google Scholar]
EMBO J. 1988; 7: 2279–2286. doi: 10.1002/j.1460-2075.1988.tb03070.x. [Статья бесплатно PMC] [PubMed] [CrossRef] [Google Scholar]
58. Koubaa S., Bremer A., Hincha D.K., Brini F. Структурные свойства и функция стабилизации фермента внутренне неупорядоченного белка LEA_4 TdLEA3 из пшеницы. науч. Отчет 2019;9:3720. doi: 10.1038/s41598-019-39823-w. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
59. Ковач Д., Кальмар Э., Торок З., Томпа П. Шаперонная активность ERD10 и ERD14 , двух неупорядоченных белков растений, связанных со стрессом. Завод Физиол. 2008; 147:381. doi: 10.1104/стр.108.118208. [Статья PMC бесплатно] [PubMed] [CrossRef] [Google Scholar]
Консервативные глицины контролируют нарушения и функционируют в холоднорегулируемом белке COR15A. Биомолекулы. 2019;9:84. дои: 10.3390/биом84. [Статья бесплатно PMC] [PubMed] [CrossRef] [Google Scholar]
61. Хомчински П., Сакки Н. Одноэтапный метод выделения РНК путем экстракции кислотным тиоцианатом гуанидиния-фенолом-хлороформом: двадцать с чем-то лет спустя. Нац. протокол 2006; 1: 581–585. doi: 10.1038/nprot.2006.83. [PubMed] [CrossRef] [Google Scholar]
Нац. протокол 2006; 1: 581–585. doi: 10.1038/nprot.2006.83. [PubMed] [CrossRef] [Google Scholar]
62. Ли З., Трик Х. Н. Быстрый метод высококачественного выделения РНК из эндосперма семян, содержащего высокий уровень крахмала. БиоТехники. 2005; 38: 872–876. дои: 10.2144/05386BM05. [PubMed] [CrossRef] [Академия Google]
63. До П.Т., Дегенкольбе Т., Эрбан А., Хейер А.Г., Копка Дж., Коль К.И., Хинча Д.К., Зутер Э. Анализ метаболизма полиаминов риса в условиях контролируемого длительного стресса засухи. ПЛОС ОДИН. 2013;8:e60325. doi: 10.1371/journal.pone.0060325. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
64. Li H. Minimap2: Попарное выравнивание нуклеотидных последовательностей. Биоинформатика. 2018;34:3094–3100. doi: 10.1093/биоинформатика/bty191. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
65. Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R. Формат выравнивания/карты последовательностей и SAMtools. Биоинформатика. 2009;25:2078–2079. doi: 10.1093/биоинформатика/btp352. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
Биоинформатика. 2009;25:2078–2079. doi: 10.1093/биоинформатика/btp352. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
66. Tseng E. CDNA Cupcake. [(по состоянию на 29 ноября 2019 г.)]; Доступно в Интернете: https://github.com/Magdoll/cDNA_Cupcake
67. Ценг Э. Коджент. [(по состоянию на 29 ноября 2019 г.)]; Доступно онлайн: https://github.com/Magdoll/Cogent
68. Ценг Э. Когент Учебник. [(по состоянию на 29 ноября 2019 г.)]; Доступно в Интернете: https://github.com/Magdoll/Cogent/wiki/Tutorial%3A-Using-Cogent-to-collapse-redundant-transcripts-in-absence-of-genome
69. Tseng E. cDNA cupcake Wiki . [(по состоянию на 29 ноября 2019 г.)]; Доступно в Интернете: https://github.com/Magdoll/cDNA_Cupcake/wiki
70. Куинлан А.Р., Холл И.М. BEDTools: гибкий набор утилит для сравнения геномных признаков. Биоинформатика. 2010; 26:841–842. дои: 10.1093/биоинформатика/btq033. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
71. Figshare. [(по состоянию на 30 октября 2020 г.)]; Доступно на сайте: www.figshare.com
[(по состоянию на 30 октября 2020 г.)]; Доступно на сайте: www.figshare.com
72. Уотерхаус Р.М., Сеппи М., Симао Ф.А., Манни М., Иоаннидис П., Ключников Г., Кривенцева Е.В., Здобнов Е.М. Приложения BUSCO от оценки качества до предсказания генов и филогеномики. Мол. биол. Эвол. 2018; 35: 543–548. doi: 10.1093/molbev/msx319. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
73. Lee TH, Guo H., Wang X., Kim C., Paterson A.H. SNPhylo: Конвейер для построения филогенетического дерева на основе огромных данных SNP. БМС Геном. 2014;15:1471. дои: 10.1186/1471-2164-15-162. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
74. Li H. Статистическая основа для определения SNP, обнаружения мутаций, сопоставления ассоциаций и оценки генетических параметров популяции на основе данных секвенирования. Биоинформатика. 2011;27:2987–2993. doi: 10.1093/биоинформатика/btr509. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
75. Рамбо А. FigTree v1. 4. [(по состоянию на 29 ноября 2019 г.)]; Доступно в Интернете: http://tree.bio.ed.ac.uk/software/figtree/
4. [(по состоянию на 29 ноября 2019 г.)]; Доступно в Интернете: http://tree.bio.ed.ac.uk/software/figtree/
76. Pertea G., Pertea M. GFF Utilities: GffRead и GffCompare [версия 1; экспертная оценка: одобрено 3] F1000Research. 2020;9:304. doi: 10.12688/f1000research.23297.1. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
77. Haas B.J., Papanicolaou A., Yassour M., Grabherr M., Blood PD, Bowden J., Couger M.B., Eccles D., Li B. ., Либер М. и др. Реконструкция последовательности транскрипта De novo из секвенирования РНК с использованием платформы Trinity для создания эталонов и анализа. Нац. протокол 2013; 8: 1494–1512. doi: 10.1038/nprot.2013.084. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
78. Консорциум UniProt, UniProt: Всемирный центр знаний о белках. Нуклеиновые Кислоты Res. 2018;47:D506–D515. [Бесплатная статья PMC] [PubMed] [Google Scholar]
79. Эдди С. Скрытые марковские модели. [(по состоянию на 19 ноября 2019 г. )]; Доступно в Интернете: http://hmmer.org/
)]; Доступно в Интернете: http://hmmer.org/
80. Эль-Гебали С., Мистри Дж., Бейтман А., Эдди С.Р., Лучани А., Поттер С.К., Куреши М., Ричардсон Л.Дж., Салазар Г.А., Смарт А. и др. База данных семейств белков Pfam в 2019 г. Nucleic Acids Res. 2019;47:D427–D432. doi: 10.1093/nar/gky995. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
81. Брайант Д.М., Джонсон К., ДиТоммазо Т., Тикл Т., Кугер М.Б., Пайзин-Догру Д., Ли Т.Дж., Ли Н.Д., Куо Т.Х., Дэвис Ф.Г. и др. Тканевой картированный транскриптом аксолотля de novo позволяет идентифицировать факторы регенерации конечностей. Cell Rep. 2017; 18: 762–776. doi: 10.1016/j.celrep.2016.12.063. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
82. Усадель Б. Веб-инструмент Mercator4. [(по состоянию на 30 марта 2020 г.)]; Доступно на сайте: https://plabipd.de/portal/mercator4
83. EnsemblPlants, Oryza Wildspecies. [(по состоянию на 2 апреля 2019 г.)]; Доступно в Интернете: https://plants. ensembl.org/index.html
ensembl.org/index.html
84. Conway J.R., Lex A., Gehlenborg N. UpSetR: пакет R для визуализации пересекающихся множеств и их свойств. Биоинформатика. 2017;33:2938–2940. doi: 10.1093/биоинформатика/btx364. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
85. Брей Н.Л., Пиментел Х., Мелстед П., Пахтер Л. Почти оптимальная вероятностная количественная оценка секвенирования РНК. Нац. Биотехнолог. 2016; 34: 525–527. doi: 10.1038/nbt.3519. [PubMed] [CrossRef] [Google Scholar]
86. Лав М.И., Хубер В., Андерс С. Модерированная оценка изменения кратности и дисперсии для данных секвенирования РНК с помощью DESeq2. Геном биол. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
87. Wickham H. ggplot2: Elegant Graphics for Data Analysis. Спрингер; Нью-Йорк, штат Нью-Йорк, США: 2016. [Google Scholar]
88. Кассамбара А. Пакет Ggpubr R: готовые к публикации графики на основе Ggplot2. [(по состоянию на 30 апреля 2019 г. )]; Доступно в Интернете: https://github.com/kassambara/ggpubr
)]; Доступно в Интернете: https://github.com/kassambara/ggpubr
89. Оги Б. GridExtra: Различные функции для «сетчатой» графики. Пакет R версии 2.3. [(по состоянию на 30 апреля 2019 г.)]; Доступно в Интернете: http://CRAN.R-project.org/package=gridExtra
90. Уикхем Х. Изменение формы данных с помощью пакета reshape. Дж. Стат. ПО 2007; 21:1–20. doi: 10.18637/jss.v021.i12. [Перекрестная ссылка] [Академия Google]
91. Эдгар Р., Домрачев М., Лаш А.Е. Омнибус экспрессии генов: хранилище данных массива экспрессии генов и гибридизации NCBI. Нуклеиновые Кислоты Res. 2002;30:2074. doi: 10.1093/нар/30.1.207. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
Полноразмерный транскриптом мРНК de novo, сгенерированный из гибридно-скорректированных лонг-ридов PacBio, улучшает аннотацию транскрипта и идентифицирует тысячи новых вариантов сплайсинга у атлантического лосося
Введение
Атлантический лосось ( Salmo Salar ) представляет собой вид, имеющий значительную экономическую и научную ценность. Это важный вид аквакультуры, и здесь также ведется значительный коммерческий вылов дикого лосося (ФАО, 2018). Оба этих вида деятельности значительно выигрывают от расширения знаний о генетике лосося, помогая в разведении для повышения выхода, качества и благополучия выращиваемого лосося, а также в мониторинге здоровья диких популяций (Yanez et al., 2014; Abdelrahman et al., 2017; Хьюстон и Маккуин, 2019 г.).
Это важный вид аквакультуры, и здесь также ведется значительный коммерческий вылов дикого лосося (ФАО, 2018). Оба этих вида деятельности значительно выигрывают от расширения знаний о генетике лосося, помогая в разведении для повышения выхода, качества и благополучия выращиваемого лосося, а также в мониторинге здоровья диких популяций (Yanez et al., 2014; Abdelrahman et al., 2017; Хьюстон и Маккуин, 2019 г.).
Лососевые подверглись относительно недавнему событию полной дупликации генома (WGD) (четвертая специфическая для лососевых дупликация генома позвоночных, Ss4R) примерно 80 миллионов лет назад (Allendorf and Thorgaard, 1984; Macqueen and Johnston, 2014). В настоящее время они подвергаются редиплоидизации, что делает атлантического лосося модельным видом, полезным для изучения явлений после WGD, таких как редиплоидизация и сохранение частичной тетрасомии (Lien et al., 2016; Campbell et al., 2019). Смолтификация, процесс, посредством которого атлантический лосось и другие анадромные лососевые адаптируются от жизни в пресной воде к жизни в соленой воде (Hoar, 19). 88), представляет собой еще одну интересную с научной точки зрения и уникальную трансформацию развития, которую было бы интересно изучить с помощью омик-технологий. Этот переход также представляет собой управленческую проблему в аквакультуре из-за высокого уровня смертности, связанного с периодом переноса соленой воды (Hjeltnes et al., 2019). Инфекционные заболевания, вызываемые различными патогенами, также представляют собой серьезную проблему для отрасли аквакультуры и продолжают приводить к большим экономическим потерям и ухудшению здоровья рыб (Hjeltnes et al., 2019).). Высококачественные транскриптомные ресурсы чрезвычайно ценны при изучении лежащих в основе молекулярных процессов, управляющих такими трансформациями развития, молекулярных деталей инфекционных заболеваний, а также при изучении явлений после WGD. Они также являются очень важными ресурсами для постоянного управления аквакультурой, основанного на знаниях, для улучшения благополучия рыбы и обеспечения роста отрасли аквакультуры (Abdelrahman et al.
88), представляет собой еще одну интересную с научной точки зрения и уникальную трансформацию развития, которую было бы интересно изучить с помощью омик-технологий. Этот переход также представляет собой управленческую проблему в аквакультуре из-за высокого уровня смертности, связанного с периодом переноса соленой воды (Hjeltnes et al., 2019). Инфекционные заболевания, вызываемые различными патогенами, также представляют собой серьезную проблему для отрасли аквакультуры и продолжают приводить к большим экономическим потерям и ухудшению здоровья рыб (Hjeltnes et al., 2019).). Высококачественные транскриптомные ресурсы чрезвычайно ценны при изучении лежащих в основе молекулярных процессов, управляющих такими трансформациями развития, молекулярных деталей инфекционных заболеваний, а также при изучении явлений после WGD. Они также являются очень важными ресурсами для постоянного управления аквакультурой, основанного на знаниях, для улучшения благополучия рыбы и обеспечения роста отрасли аквакультуры (Abdelrahman et al. , 2017).
, 2017).
Набор хромосом Salmo Salar 9Геном 0497 стал общедоступным с 2015 года благодаря усилиям Международного сотрудничества по секвенированию генома атлантического лосося (ICSASG) (Lien et al., 2016), но ресурсы на уровне транскриптов ограничены. Была проведена некоторая работа по созданию полноразмерных транскриптов мРНК для Salmo Salar (Andreassen et al., 2009; Leong et al., 2010). Однако подавляющее большинство транскриптов, кодирующих белки, в базе данных NCBI RefSeq были аннотированы с использованием in silico 9.0497 предсказаний на основе последовательности генома, поддерживаемых и скорректированных с помощью EST, полученных в результате секвенирования библиотек кДНК и высокопроизводительного секвенирования (HTS) транскриптомов на платформах, производящих короткие данные (на момент написания 498 177 EST и 4 475 852 530 коротких читает) (Hagen-Larsen et al., 2005; Adzhubei et al., 2007; Koop et al., 2008; NCBI, 2015). Хотя эти методы полезны для определения присутствия определенных генных продуктов, они гораздо менее полезны для характеристики изоформ транскриптов (Conesa et al. , 2016). Таким образом, идентификация вариантов сплайсинга и возможное неправильное размещение коротких последовательностей транскриптов в геноме из-за существования очень похожих антропологических генов (в результате специфичной для лососевых WGD) представляют собой проблемы, которые нелегко решить у лососевых, если полагаться только на секвенирование коротких транскриптов. (Леонг и др., 2010; Лю и др., 2012). Полноразмерный кодирующий белок транскриптом вида (CDS, а также 5′- и 3’UTR) и его репертуар вариантов сплайсинга является важным ресурсом для надежной аннотации транскриптов, кодирующих белок, и понимания того, как такие структурные варианты влияют на болезнь и экономически важные признаки сельскохозяйственных животных (Abdelrahman et al., 2017; Giuffra et al., 2019).
, 2016). Таким образом, идентификация вариантов сплайсинга и возможное неправильное размещение коротких последовательностей транскриптов в геноме из-за существования очень похожих антропологических генов (в результате специфичной для лососевых WGD) представляют собой проблемы, которые нелегко решить у лососевых, если полагаться только на секвенирование коротких транскриптов. (Леонг и др., 2010; Лю и др., 2012). Полноразмерный кодирующий белок транскриптом вида (CDS, а также 5′- и 3’UTR) и его репертуар вариантов сплайсинга является важным ресурсом для надежной аннотации транскриптов, кодирующих белок, и понимания того, как такие структурные варианты влияют на болезнь и экономически важные признаки сельскохозяйственных животных (Abdelrahman et al., 2017; Giuffra et al., 2019).
Секвенирование с длинным считыванием на основе технологии секвенирования PacBio Iso-Seq SMRT позволяет получать полноразмерные последовательности транскриптов (например, полноразмерные мРНК) путем секвенирования отдельных молекул (Rhoads and Au, 2015). Этот метод решает проблемы, связанные со сборкой данных HTS с коротким считыванием, путем создания чтений, которые охватывают весь транскрипт, кодирующий белок, включая CDS и 5′- и 3’UTR мРНК. Таким образом, этот метод позволяет точно идентифицировать различные варианты сплайсинга, а у лососевых позволяет различать транскрипты с очень похожими антропологическими кодирующими последовательностями, поскольку полное чтение (включая менее консервативный 3’UTR) генерируется из одной молекулы. . Возможность однозначно охарактеризовать 3’UTR конкретных вариантов транскриптов также будет полезна для изучения регуляторных элементов, нацеленных на эти области, таких как микроРНК (Woldemariam et al., 2019)., 2020; Шве и др., 2020). Однако подавляющее большинство 3’UTR в настоящем атлантическом эталонном транскриптоме (RefSeq) (NCBI, 2015) предсказано на основе последовательности генома атлантического лосося с поддержкой короткого чтения (записи RefSeq XM).
Этот метод решает проблемы, связанные со сборкой данных HTS с коротким считыванием, путем создания чтений, которые охватывают весь транскрипт, кодирующий белок, включая CDS и 5′- и 3’UTR мРНК. Таким образом, этот метод позволяет точно идентифицировать различные варианты сплайсинга, а у лососевых позволяет различать транскрипты с очень похожими антропологическими кодирующими последовательностями, поскольку полное чтение (включая менее консервативный 3’UTR) генерируется из одной молекулы. . Возможность однозначно охарактеризовать 3’UTR конкретных вариантов транскриптов также будет полезна для изучения регуляторных элементов, нацеленных на эти области, таких как микроРНК (Woldemariam et al., 2019)., 2020; Шве и др., 2020). Однако подавляющее большинство 3’UTR в настоящем атлантическом эталонном транскриптоме (RefSeq) (NCBI, 2015) предсказано на основе последовательности генома атлантического лосося с поддержкой короткого чтения (записи RefSeq XM).
Разработка технологии секвенирования PacBio Iso-Seq SMRT позволила проводить высокопроизводительное секвенирование с длительным считыванием, подходящее для секвенирования полного транскриптома вида или создания тканеспецифических транскриптомов для изучения экспрессии тканеспецифических генов (Wang et al. , 2016). ). Более высокому уровню ошибок, связанному с секвенированием с длинным считыванием, можно противодействовать путем создания консенсуса из нескольких чтений одной молекулы (чтения высокого качества) с применением метода Iso-Seq (Gordon et al., 2015; Rhoads and Au, 2015). Частота ошибок может быть дополнительно снижена с помощью гибридных методов коррекции ошибок на основе графов (Au et al., 2012; Salmela and Rivals, 2014; Sahraeian et al., 2017). Этот подход использует длинные чтения в качестве основы для выравнивания коротких чтений, которые включают исправление ошибок последовательности. Таким образом, длинные считывания предоставляют структурную информацию обо всех изоформах, в то время как изоформы длинных чтений исправляются с исправлением ошибок за счет гораздо большего количества коротких чтений с превосходной точностью считывания. Это позволяет получить de novo с качеством, сравнимым с любым справочным ресурсом, без использования текущей сборки генома в качестве источника исправления ошибок (Feng et al.
, 2016). ). Более высокому уровню ошибок, связанному с секвенированием с длинным считыванием, можно противодействовать путем создания консенсуса из нескольких чтений одной молекулы (чтения высокого качества) с применением метода Iso-Seq (Gordon et al., 2015; Rhoads and Au, 2015). Частота ошибок может быть дополнительно снижена с помощью гибридных методов коррекции ошибок на основе графов (Au et al., 2012; Salmela and Rivals, 2014; Sahraeian et al., 2017). Этот подход использует длинные чтения в качестве основы для выравнивания коротких чтений, которые включают исправление ошибок последовательности. Таким образом, длинные считывания предоставляют структурную информацию обо всех изоформах, в то время как изоформы длинных чтений исправляются с исправлением ошибок за счет гораздо большего количества коротких чтений с превосходной точностью считывания. Это позволяет получить de novo с качеством, сравнимым с любым справочным ресурсом, без использования текущей сборки генома в качестве источника исправления ошибок (Feng et al.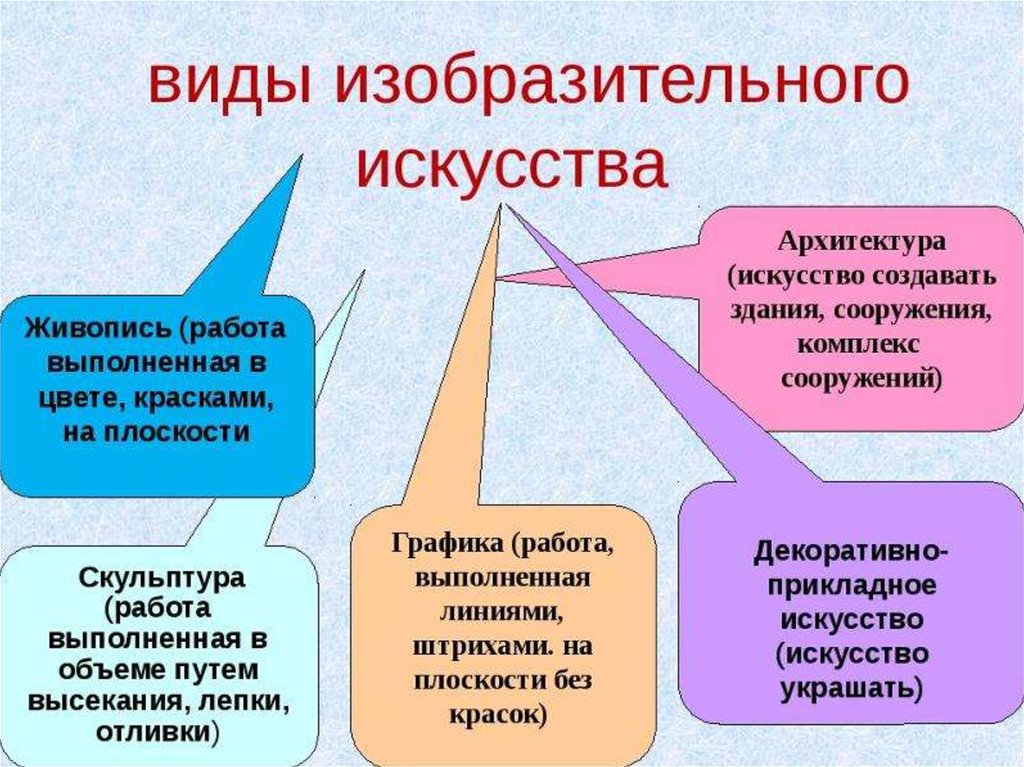 , 2019). Это особенно важно для немодельных видов, где сборки генома в целом имеют значительный потенциал для улучшения качества.
, 2019). Это особенно важно для немодельных видов, где сборки генома в целом имеют значительный потенциал для улучшения качества.
Цель этого исследования состояла в том, чтобы предоставить первый высококачественный полноразмерный ресурс транскриптома, кодирующий белок, для атлантического лосося. Нас особенно интересует изучение изменений экспрессии и регуляции экспрессии генов во время смолтификации и переноса морской водой, а также изменений экспрессии и регуляции генов в ответ на инфекционные заболевания (Woldemariam et al., 2019)., 2020; Шве и др., 2020). Таким образом, два образца, включенных в это исследование, были выбраны из контрольного исследования для выявления полноразмерных последовательностей мРНК, экспрессируемых в голове и почках при инфицировании альфа-вирусом лососевых (SAV) (McLoughlin and Graham, 2007; Andreassen et al., 2017; Бернхардт и др., 2021). Были выбраны образцы из головы и почки, так как это один из основных иммунных органов у рыб, который часто используется в иммунологических исследованиях экспрессии генов у рыб (Bjørgen and Koppang, 2021). Образцы трех различных основных стадий плавления; предсмолт, смолтифицированную рыбу и после пересадки в морскую воду отбирали из жабр, печени и головы-почки. Все эти органы являются важными органами в этом переходном этапе развития, и образцы были взяты из нашего недавнего и продолжающегося исследования смолтификации (Shwe et al., 2020). Другой целью была разработка и оценка конвейера транскриптома на основе длинных считываний. Мы использовали комбинацию существующих инструментов для анализа последовательностей, курирования и аннотирования данных транскриптов PacBio Iso-Seq, применяя платформы для сиквела I и сиквела II. Затем качество длинных ридов с платформы PacBio было улучшено за счет использования дополнительных данных расшифровки, сгенерированных на платформе кратких ридов Illumina. Применение гибридных алгоритмов исправления ошибок, дополненных скриптами собственной разработки, позволило повысить точность последовательности. Наконец, после создания высококачественного набора данных транскриптома, состоящего из полноразмерных мРНК с полными CDS, транскрипты были функционально аннотированы.
Образцы трех различных основных стадий плавления; предсмолт, смолтифицированную рыбу и после пересадки в морскую воду отбирали из жабр, печени и головы-почки. Все эти органы являются важными органами в этом переходном этапе развития, и образцы были взяты из нашего недавнего и продолжающегося исследования смолтификации (Shwe et al., 2020). Другой целью была разработка и оценка конвейера транскриптома на основе длинных считываний. Мы использовали комбинацию существующих инструментов для анализа последовательностей, курирования и аннотирования данных транскриптов PacBio Iso-Seq, применяя платформы для сиквела I и сиквела II. Затем качество длинных ридов с платформы PacBio было улучшено за счет использования дополнительных данных расшифровки, сгенерированных на платформе кратких ридов Illumina. Применение гибридных алгоритмов исправления ошибок, дополненных скриптами собственной разработки, позволило повысить точность последовательности. Наконец, после создания высококачественного набора данных транскриптома, состоящего из полноразмерных мРНК с полными CDS, транскрипты были функционально аннотированы.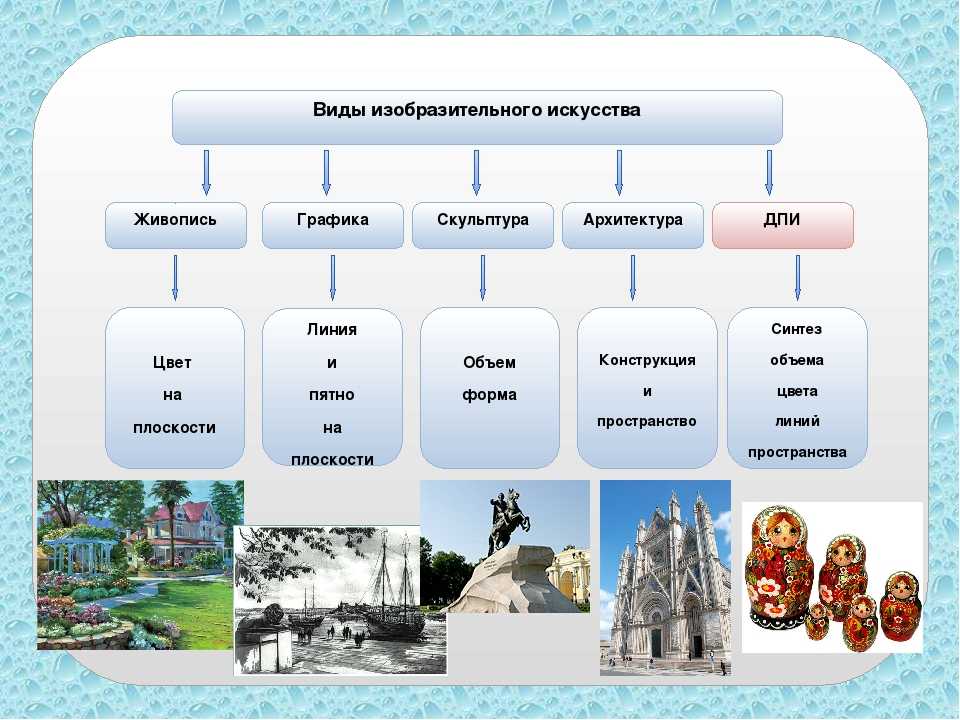 Конвейерная обработка не зависит от последовательности генома атлантического лосося и других источников транскриптов, таких как RefSeq для исправления ошибок. Это позволило экспериментально проверить транскрипты, ранее предсказанные на основе последовательности генома и EST, с помощью долгочитаемого de novo , а также новые сплайс-варианты и паралоги, подлежащие надежной характеристике.
Конвейерная обработка не зависит от последовательности генома атлантического лосося и других источников транскриптов, таких как RefSeq для исправления ошибок. Это позволило экспериментально проверить транскрипты, ранее предсказанные на основе последовательности генома и EST, с помощью долгочитаемого de novo , а также новые сплайс-варианты и паралоги, подлежащие надежной характеристике.
Материалы и методы
Материалы для образцов рыбы
В таблице 1 представлен обзор секвенированных образцов, включая информацию об экспериментальных условиях и типе их органов. В таблице также указаны уникальные метки, используемые для каждого образца в последующем анализе. Два образца головы и почек, включенные в исследование (SAV_Control и SAV_challenge, таблица 1), были получены от одной здоровой контрольной рыбы и одной рыбы, зараженной альфавирусом лососевых, соответственно. Пробное испытание было проведено в Промышленной и водной лаборатории (ILAB, Бергенский центр высоких технологий, Берген, Норвегия) в феврале/марте 2018 г. (Bernhardt et al., 2021). После смолта рыба породы SF Optimal (Stofnfiskur Iceland) подверглась заражению в результате сожительства в соленой воде с линьками лосося (рыбой-носителем), которым инъецировали альфавирус лосося подтипа 3 (SAV3) из Норвегии (Taksdal et al., 2015). Все рыбы, использованные в испытании с контрольным заражением, были невакцинированы, что позволяло изучать иммунный ответ после вирусной инфекции SAV. Все рыбы дали отрицательный результат на SAV3, вирус инфекционной анемии лосося (ISAV), вирус инфекционного панкреонекроза (IPNV), вирус миокардита рыб (PMCV), ортореовирус рыб (PRV) и вирус оспы жабер лосося (SGPV) до испытания с контрольным заражением, подтверждающим, что контрольная рыба была здоровой рыбой, не зараженной каким-либо рыбным вирусом, обычно встречающимся в аквакультуре. Средний вес рыбы составил 110,9 г.г, а в опытный период содержание растворенного кислорода составляло 79–97%, температура воды – 11,5–12,4 °С, соленость – 34,1–34,5‰ по всем резервуарам.
(Bernhardt et al., 2021). После смолта рыба породы SF Optimal (Stofnfiskur Iceland) подверглась заражению в результате сожительства в соленой воде с линьками лосося (рыбой-носителем), которым инъецировали альфавирус лосося подтипа 3 (SAV3) из Норвегии (Taksdal et al., 2015). Все рыбы, использованные в испытании с контрольным заражением, были невакцинированы, что позволяло изучать иммунный ответ после вирусной инфекции SAV. Все рыбы дали отрицательный результат на SAV3, вирус инфекционной анемии лосося (ISAV), вирус инфекционного панкреонекроза (IPNV), вирус миокардита рыб (PMCV), ортореовирус рыб (PRV) и вирус оспы жабер лосося (SGPV) до испытания с контрольным заражением, подтверждающим, что контрольная рыба была здоровой рыбой, не зараженной каким-либо рыбным вирусом, обычно встречающимся в аквакультуре. Средний вес рыбы составил 110,9 г.г, а в опытный период содержание растворенного кислорода составляло 79–97%, температура воды – 11,5–12,4 °С, соленость – 34,1–34,5‰ по всем резервуарам. Образцы (зараженные рыбы и контроль) собирали на 37-й день в испытании с заражением SAV и позже фиксировали в РНК (Life Technologies, Карлсбад, Калифорния, США) сразу после сбора. Успешная инфекция SAV3 была подтверждена обнаружением вирусной последовательности в контрольном образце. Экспериментальное исследование было одобрено Национальным исследовательским органом Норвегии (NARA). Весь лосось, использованный для отбора проб в эксперименте, перед отбором проб был подвергнут эвтаназии в соответствии со стандартными протоколами, утвержденными Норвежским управлением по безопасности пищевых продуктов. Для простоты эти две выборки называются выборками SAV.
Образцы (зараженные рыбы и контроль) собирали на 37-й день в испытании с заражением SAV и позже фиксировали в РНК (Life Technologies, Карлсбад, Калифорния, США) сразу после сбора. Успешная инфекция SAV3 была подтверждена обнаружением вирусной последовательности в контрольном образце. Экспериментальное исследование было одобрено Национальным исследовательским органом Норвегии (NARA). Весь лосось, использованный для отбора проб в эксперименте, перед отбором проб был подвергнут эвтаназии в соответствии со стандартными протоколами, утвержденными Норвежским управлением по безопасности пищевых продуктов. Для простоты эти две выборки называются выборками SAV.
Таблица 1. Распределение типов тканей и экспериментальных условий для образцов, секвенированных с метками образцов, использованными в окончательном наборе данных.
Девять оставшихся включенных образцов были собраны у рыб, использовавшихся в исследовании изменений экспрессии генов миРНК во время смолтификации и раннего периода соленой воды (Shwe et al. , 2020). Образцы были взяты из головы, почек, жабр и печени до смолтификации (0-й день; HKU1, GiU1, LiU1), в конце смолтификации (81-й день; HKU4, GiU4, LiU4) и через 4 недели после переноса морской воды. (день 111; HKU7, GiU7, LiU7). Образцы печени и жабр с 0-го дня были взяты у одной рыбы, а образец головы и почек — у другой. Три образца на 81-й день были получены от одной и той же рыбы, и то же самое относится и к трем образцам на 111-й день. Подробная информация об условиях смолтификации и сборе образцов приведена в Shwe et al. (2020). Вскоре перед отбором проб рыбу анестезировали передозировкой MS-222 (метансульфонат трикаина 123, 0,1 г/л) и убивали ударом по голове. Образцы тканей немедленно собирали, замораживали в жидком водороде и хранили при температуре -80°С. Все процедуры обращения с рыбой соответствовали директивам законодательства ЕС (2010/63/EU), а также норвежскому законодательству. Эксперимент считался нерегулируемой процедурой в соответствии с Национальным законодательством об исследованиях на животных, поскольку рыба не подвергалась какой-либо боли или стрессу.
, 2020). Образцы были взяты из головы, почек, жабр и печени до смолтификации (0-й день; HKU1, GiU1, LiU1), в конце смолтификации (81-й день; HKU4, GiU4, LiU4) и через 4 недели после переноса морской воды. (день 111; HKU7, GiU7, LiU7). Образцы печени и жабр с 0-го дня были взяты у одной рыбы, а образец головы и почек — у другой. Три образца на 81-й день были получены от одной и той же рыбы, и то же самое относится и к трем образцам на 111-й день. Подробная информация об условиях смолтификации и сборе образцов приведена в Shwe et al. (2020). Вскоре перед отбором проб рыбу анестезировали передозировкой MS-222 (метансульфонат трикаина 123, 0,1 г/л) и убивали ударом по голове. Образцы тканей немедленно собирали, замораживали в жидком водороде и хранили при температуре -80°С. Все процедуры обращения с рыбой соответствовали директивам законодательства ЕС (2010/63/EU), а также норвежскому законодательству. Эксперимент считался нерегулируемой процедурой в соответствии с Национальным законодательством об исследованиях на животных, поскольку рыба не подвергалась какой-либо боли или стрессу. Таким образом, этот эксперимент не требовал одобрения Норвежского управления по безопасности пищевых продуктов. Для простоты эти образцы называются образцами смолтификации.
Таким образом, этот эксперимент не требовал одобрения Норвежского управления по безопасности пищевых продуктов. Для простоты эти образцы называются образцами смолтификации.
Подготовка библиотеки PacBio и секвенирование Iso-Seq, подготовка библиотеки Illumina и секвенирование РНК
Все девять смолтифицированных образцов были обработаны в Институте Эрлхема (Норвич, Англия). Экстракцию РНК проводили с использованием мини-набора Qiagen RNeasy (Qiagen, Hilden, Germany) с расщеплением ДНКазы на колонке с использованием набора ДНКазы без РНКазы в соответствии с протоколом производителя. Экстракты общей РНК использовались как для секвенирования с длинным считыванием PacBio, так и для секвенирования с короткими парными концами Illumina. Для секвенирования с длительным считыванием использовали препарат PacBio неразмерной выбранной библиотеки Iso-seq. Был применен протокол Express Template Prep 2, требующий значений RIN> 8 для образцов РНК, и каждый из девяти образцов был обработан индивидуально. Полученные комплексы кДНК-адаптер из девяти образцов секвенировали на одной ячейке PacBio Sequel II 8M SMRT каждый. Для короткого секвенирования на платформе Illumina для подготовки библиотеки использовали набор автоматизированных библиотек NEBNext Ultra II Directional RNA-Seq с отбором Poly-A (New England Biolabs, Inc., Ипсвич, Массачусетс, США). -Концевое секвенирование (150 п.н.) выполняли с использованием одной проточной кюветы Illumina NovaSeq 6000 SP для всех девяти образцов, мультиплексированных вместе.
Полученные комплексы кДНК-адаптер из девяти образцов секвенировали на одной ячейке PacBio Sequel II 8M SMRT каждый. Для короткого секвенирования на платформе Illumina для подготовки библиотеки использовали набор автоматизированных библиотек NEBNext Ultra II Directional RNA-Seq с отбором Poly-A (New England Biolabs, Inc., Ипсвич, Массачусетс, США). -Концевое секвенирование (150 п.н.) выполняли с использованием одной проточной кюветы Illumina NovaSeq 6000 SP для всех девяти образцов, мультиплексированных вместе.
Экстракция РНК, подготовка библиотеки и секвенирование двух образцов SAV были выполнены Genewiz Germany GmbH (Лейпциг, Германия). РНК экстрагировали с помощью набора RNeasy Plus Mini Kit (Qiagen, Hilden, Germany) в соответствии с протоколом производителя, а образцы, использованные для секвенирования, имели RIN > 8. Набор для синтеза кДНК SMRTer PCR (Clontech Laboratories, Inc., Маунтин-Вью, Калифорния, США) без выбора размера, а комплекс кДНК-адаптер был создан с использованием набора для подготовки матрицы SMRTbell V1. 0. (Pacific Biosciences of California, Inc., Менло-Парк, Калифорния, США). Каждый образец был секвенирован с использованием PacBio Sequel I. Каждый образец был секвенирован на двух клетках 1M v2 SMRT, чтобы компенсировать меньшее количество прочтений в Sequel I по сравнению с Sequel II. Для короткого секвенирования применяли набор для подготовки библиотеки NEBnext Ultra RNA (New England Biolabs, Inc., Ипсвич, Массачусетс, США) в соответствии с протоколом производителя. Секвенирование парных концов (150 п.н.) проводили на платформе Illumina HiSeq 4000.
0. (Pacific Biosciences of California, Inc., Менло-Парк, Калифорния, США). Каждый образец был секвенирован с использованием PacBio Sequel I. Каждый образец был секвенирован на двух клетках 1M v2 SMRT, чтобы компенсировать меньшее количество прочтений в Sequel I по сравнению с Sequel II. Для короткого секвенирования применяли набор для подготовки библиотеки NEBnext Ultra RNA (New England Biolabs, Inc., Ипсвич, Массачусетс, США) в соответствии с протоколом производителя. Секвенирование парных концов (150 п.н.) проводили на платформе Illumina HiSeq 4000.
Конвейер для создания неизбыточного
de novo Ресурс транскриптома с исправленными ошибками высококачественными лонг-ридами без вкрапленных повторов Необработанные лонгриды Iso-Seq из секвенирования PacBio Iso-seq конвейер IsoSeq3 (PacBio, 2020 г.), как показано на рис. 1. Версия SMRT link 8.0 использовалась для образцов смолтификации, а 6.0 — для образцов SAV, поскольку они были секвенированы и обработаны до выпуска версии 8. 0. Данные по каждому образцу обрабатывались независимо. Только чтение высокого качества (HQ), что означает, что они поддерживались как минимум двумя FLNC и с прогнозируемой точностью последовательности ≥ 99% (> Q20) (результаты fasta, рис. 1) использовались в нашем последующем анализе.
0. Данные по каждому образцу обрабатывались независимо. Только чтение высокого качества (HQ), что означает, что они поддерживались как минимум двумя FLNC и с прогнозируемой точностью последовательности ≥ 99% (> Q20) (результаты fasta, рис. 1) использовались в нашем последующем анализе.
Рис. 1. Конвейер PacBio Isoseq3 для обработки данных SMRT-секвенирования. Каждая волна нулевой моды (ZMW) предоставляет информацию от одной ДНК-полимеразы, которая повторно секвенирует каждый адаптер кДНК-SMRTBell. Консенсус: программа CCS генерирует консенсусную последовательность для каждого чтения, которая содержит полный повторяющийся комплекс вставка-адаптер. Demulitplex: lima отфильтровывает последовательности с нежелательными комбинациями праймеров, обрезает последовательности-адаптеры и ориентирует чтения в 5’→3′-ориентации. Уточнить: программа уточнения отфильтровывает конкатемеры и последовательности без полиА-хвостов длиной не менее 20 п.н. Наконец, он обрезает хвосты полиА из оставшихся последовательностей. Кластер: Кластер Isoseq выполняет консервативную кластеризацию последовательностей и использует выравнивание частичного порядка для создания согласованной последовательности для каждого кластера. Результат классифицируется как высокое качество или низкое качество в зависимости от прогнозируемой точности. Окончательные результаты представляют собой высококачественные и низкокачественные последовательности в формате fastq. Эта цифра используется с разрешения Pacific Biosciences.
Кластер: Кластер Isoseq выполняет консервативную кластеризацию последовательностей и использует выравнивание частичного порядка для создания согласованной последовательности для каждого кластера. Результат классифицируется как высокое качество или низкое качество в зависимости от прогнозируемой точности. Окончательные результаты представляют собой высококачественные и низкокачественные последовательности в формате fastq. Эта цифра используется с разрешения Pacific Biosciences.
Cutadapt 1.18 (Martin, 2011) был применен для удаления адаптером ридов Illumina, и качество было проверено с помощью FASTQC (Andrews, 2010; рис. 2, Cutadapt). Высококачественные считывания парных концов применялись для гибридной коррекции ошибок считываний HQ, сгенерированных Iso-Seq, с использованием версии 0.9 алгоритма LoRDEC [Коррекция ошибок графа де Брейна (DBG) с длительным считыванием] (Salmela and Rivals, 2014) с Illumina считывает данные из того же образца, используя размер k-mer 21 и порог солидности 3 (рис.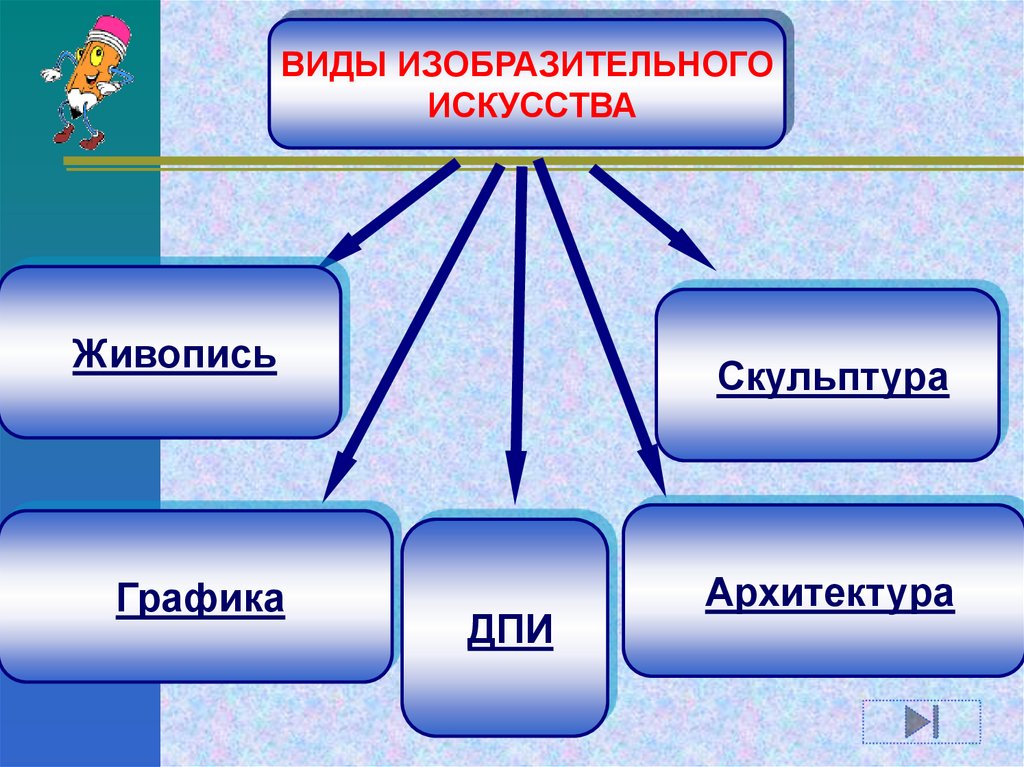 2, LoRDEC).
2, LoRDEC).
Рис. 2. Обзор конвейера анализа от обработки последовательностей до неизбыточного транскриптома высокого качества с исправлением ошибок. Чтения высокого качества PacBio SMRT были получены с платформы PacBio. Прочтения Illumina сначала обрезали с помощью cutadapt для удаления последовательностей адаптера. Впоследствии они были использованы для создания графика Де Брейна для LoRDEC для исправления ошибок считываний высокого качества для каждой выборки. Собственный скрипт Python: чтения с исправлением ошибок были отфильтрованы на основе степени поддержки Illumina и охвата операций чтения высокого качества. Repeatmasker использовался для выявления и удаления ридов, содержащих известные повторы с длинными вкраплениями. Последовательности, которые могут быть точно сопоставлены с 9Геном 0496 Salmo salar или Salmo trutta был кластеризован с использованием cdna_Cupcake, тогда как остальные последовательности были кластеризованы с использованием Cogent. Все чтения были дополнительно кластеризованы с помощью CD-Hit перед аннотацией. Конечным результатом был неизбыточный транскриптом высокого качества с исправлением ошибок.
Все чтения были дополнительно кластеризованы с помощью CD-Hit перед аннотацией. Конечным результатом был неизбыточный транскриптом высокого качества с исправлением ошибок.
Чтения HQ с исправлением ошибок (чтения EC-HQ) затем отфильтровывались с использованием внутреннего сценария Python, который удалял все чтения EC-HQ, которые менее чем на 99% покрывались диаграммой Де Брюжина, сгенерированной из чтений Illumina (внутренний сценарий python , Фигура 2). Внутренние промежутки последовательности, не поддерживаемые графом, не допускались, в то время как один процент или менее конечных концов мог не поддерживаться графом с порогом твердости 3 (строгая фильтрация со 100-процентным охватом потребовала бы не менее трех чтений Illumina для запуска). или заканчиваются у самых 5′- или 3′-концевых оснований в прочтении HQ). Одиннадцать файлов образцов с отфильтрованными считываниями EC-HQ были объединены, и каждому чтению EC-HQ был присвоен новый уникальный идентификационный номер. В то же время происхождение образца и количество поддерживающих последовательностей FLNC из IsoSeq3 для каждого нового идентификатора были отмечены в отдельном файле, чтобы сделать их происхождение образца и поддержку FLNC прослеживаемыми (рис. 2, внутренний сценарий Python). На практике исправление ошибок гарантировало, что любая пара оснований в длинных считываниях поддерживалась как минимум тремя считываниями Illumina (среднее качество phred 36), которые последовательно охватывают длинные считывания HQ. В тех случаях, когда имелись различия в положениях отдельных оснований или небольшие числовые различия в удлинении гомополимера между считыванием HQ и вспомогательными разделами графика Де Брюжина, считывания HQ фактически корректировались последовательностями Illumina.
Все соединенные чтения EC-HQ были проверены в базе данных Dfam версии 3.0 (Hubley et al., 2016) с использованием Repeatmasker версии 4.0.9 (Smit et al., 2013) как части программного пакета OmicsBox, с использованием Поисковая система RMBlast с настройками скорости и чувствительности по умолчанию (порог оценки Смита-Уотермана 225). Любое считывание EC-HQ, которое соответствовало любому из вкрапленных повторов, присутствующих у костистых рыб ( Actinopterygii ), было удалено с использованием внутреннего скрипта Python (Repeatmasker, рисунок 2).
Группировка прочтений EC-HQ, вероятно, происходящих из одного и того же локуса генома, была проведена с применением cdna_Cupcake версии 12.1.0 (Tseng, 2020a) с minimap2 версии 2.17 (Li and Birol, 2018). Чтения EC-HQ были сопоставлены со сборкой ICSASG_v2 генома атлантического лосося (RefSeq номер доступа GCF_000233375.1) с параметрами по умолчанию, кроме допуска до 10 000 п.н. выступов на 5′-конце и 1000 п.н. выступов на 3′-конце. конец. Это позволило сгруппировать любые чтения EC-HQ по координатам генома и отнести к группам транскриптов, происходящих из одного и того же локуса. Кроме того, считывания с одним и тем же паттерном сплайсинга, представляющие одну и ту же изоформу, были сгруппированы вместе, сохраняя самую длинную последовательность в качестве репрезентативной последовательности изоформ кластера. Группы кластеров и координаты картирования генома были сохранены в заголовках fasta как информация, которая использовалась нижестоящими приложениями (SQANTI3). Релаксированные 5′- и 3′-выступающие отсечения использовались для того, чтобы более короткие чтения EC-HQ, которые представляли фрагменты других полноразмерных транскриптов чтения EC-HQ, группировались вместе с полноразмерными транскриптами, представляющими эту изоформу, вместо того, чтобы быть ошибочно идентифицированными. как отдельные варианты изоформ.
Чтения EC-HQ, которые плохо отображались в геноме атлантического лосося, были сопоставлены с версией RefSeq сборки fSalTru1.1 генома Salmo trutta (RefSeq номер доступа GCF_
1165.1) с использованием cdna_Cupcake и тех же параметров. Остальные последовательности, которые не картировались ни с геномами Salmo salar , ни с геномами Salmo trutta , были сопоставлены с геномом SAV (инвентарный номер GenBank KC122923), поскольку один из отдельных образцов был инфицирован SAV, а совпадающие чтения были отброшены. .
Оставшиеся чтения EC-HQ, которые не соответствовали ни одному из геномов лососевых, были кластеризованы с использованием Cogent 6.1.0 с параметрами по умолчанию (Tseng, 2020b) (Cogent, рис. 2). Cogent создает псевдогеном, пытаясь воссоздать последовательность генома, которая может дать начало всем наблюдаемым транскриптам в наборе данных, а затем кластеризует и группирует считывания EC-HQ в семейства с помощью cdna_Cupcake. Таким образом, считывания EC-HQ, которые плохо картировались ни с одной из двух последовательностей генома, были сгруппированы в транскрипты, которые, вероятно, были структурными вариантами одного и того же гена.
Все чтения EC-HQ, как те, которые были сопоставлены с любой из последовательностей сборки генома, так и те, которые были сгруппированы Cogent, были наконец сгруппированы с помощью CD-Hit версии 4.8.1 (Li and Godzik, 2006; Fu et al. ., 2012) (CD-Hit, рис. 2). Эта окончательная кластеризация сопоставила любое более короткое чтение EC-HQ с более длинной идентичной изоформой, если она присутствует в наборе данных. Настройки, используемые для выравнивания CD-попаданий, были следующими:
1. Порог идентичности последовательности 0,99.
2. Локальное выравнивание последовательностей.
3. Кластерные чтения для наиболее похожих более длинных чтений EC-HQ, если имеется более одного подходящего критерия выравнивания.
4. Короткое чтение EC-HQ должно на 99% совпадать с более длинным (1% «непокрытых» оснований). Если более короткое чтение EC-HQ превышало 3000 п.н., ограничение в 1% заменяли непокрытыми основаниями длиной 30 п.н. или менее.
5. Одно длинное чтение EC-HQ, с которым были выровнены другие более короткие чтения, могло иметь любой выступ.
Эта окончательная кластеризация гарантировала, что идентичные структурные варианты были выровнены в одно репрезентативное чтение EC-HQ. В большинстве случаев те, которые выровнены с более длинными чтениями EC-HQ, будут 5′-неполными чтениями EC-HQ или чтениями EC-HQ с неполным 3’UTR из-за неправильного праймирования в синтезе кДНК. Это гарантировало, что каждая отдельная структурная изоформа была представлена одним полноразмерным чтением EC-HQ. Собственный скрипт Python использовался для определения поддержки FLNC и предоставления образцов для каждого неизбыточного чтения EC-HQ после окончательной кластеризации. Этот неизбыточный транскриптом чтения EC-HQ был дополнительно проанализирован путем классификации структурных вариантов (Материалы и методы, «Классификация структурных вариантов») и функциональной аннотации (Материалы и методы, «Функциональная аннотация»).
Классификация структурных вариантов
Прочтения EC-HQ, которые были сгруппированы cDNA_cupcake с использованием генома Salmo salar или Salmo trutta , были классифицированы и сопоставлены с существующей аннотацией для соответствующих геномов. Этот анализ был проведен с помощью SQANTI3 (v 1.0.0) с параметрами по умолчанию (Tardaguila et al., 2018) (SQANTI3, рис. 3). SQANTI3 сравнивает каждое чтение EC-HQ с информацией аннотации генома в локусе, где он отображается. Основываясь на том, как чтения соответствуют аннотации генома, сравнения могут дать следующую классификацию основных категорий каждого чтения EC-HQ:
Рис. 3. Обзор процесса аннотирования. Транскрипты, которые были сгруппированы с использованием геномов Salmo salar или Salmo trutta , были охарактеризованы по аннотации генома для соответствующих видов с использованием SQANTI3. Все последовательности также использовались для прогнозирования открытых рамок считывания с помощью Transdecoder. Последовательности, которые, как было предсказано, содержат полную кодирующую последовательность, подвергали взрывной обработке в базе данных белков RefSeq и выполняли поиск в базе данных Interpro для получения названий генов и функциональной аннотации. Чтения были отфильтрованы на основе SQANTI-классификации, предсказания открытой рамки считывания и поддержки в данных секвенирования PacBio. Информация из пути структурной классификации и пути функциональной аннотации была добавлена к окончательному отфильтрованному транскриптому мРНК.
Полное соответствие сплайсинга (FSM), идентичное совпадению с изоформой транскрипта, представленной в аннотации генома, со всеми теми же соединениями сплайсинга и экзонами.
Incomplete Splice Match (ISM), представляющий собой неполное, но в остальном идентичное совпадение с известной изоформой. Все существующие соединения сплайсинга совпадают, но есть экзоны, отсутствующие на одном или обоих концах транскрипта EC-HQ.
Роман в каталоге (NIC), новая изоформа с комбинацией экзонов, отличной от изоформ, аннотированных в геноме, но с комбинацией известных соединений сплайсинга из ранее аннотированных изоформ.
Новая изоформа, отсутствующая в каталоге (NNC), новая изоформа, содержащая по крайней мере одно сплайс-соединение, не указанное в аннотации. Следовательно, они имеют по крайней мере один новый экзон.
Межгенный, означающий новый транскрипт, который картируется в локусе без ранее аннотированных генов в текущей версии генома атлантического лосося.
Генный интрон, означающий, что транскрипт полностью расположен внутри интрона аннотированного гена в геноме атлантического лосося.
Генный геномный, что означает, что транскрипт перекрывает аннотированные интроны и экзоны.
Антисмысловой, что означает отсутствие аннотированного гена в локусе на цепи, где совпадает транскрипт, но есть ген на обратной цепи.
Слияние, что означает, что чтение охватывает два разных аннотированных локуса в текущей аннотации генома.
Последовательности не были сопоставлены с геномом Salmo trutta , если они были успешно сопоставлены с геномом Salmo salar . Структурная классификация, указывающая, что EC-HQ относится к любой другой категории, кроме FSM в 9Таким образом, 0496 Salmo salar не исключает, что в Salmo trutta было бы нанесено другое изображение (например, FSM).
SQANTI также предоставляет дополнительную полезную информацию, такую как количество экзонов, сигналы соединения сплайсинга (канонические или нет), длину CDS, полиА-сигналы выше по течению на 3′-конце и геномный процент А ниже по течению от 3′-сайта терминации (используется для оценки был ли неправильный запуск синтеза кДНК).
Функциональная аннотация
Все прочтения EC-HQ в окончательном неизбыточном наборе данных транскриптома (рис. 2) были подвергнуты функциональной аннотации с использованием программного пакета OmicsBox (рис. 3; BioBam, 2019 г.).). Кодирующие последовательности были предсказаны с помощью TransDecoder v5.5.0. (Haas et al., 2013; Haas and Papanicolaou, 2015) с использованием поиска по гомологии в отношении Pfam 32 для подтверждения ORF с минимальной длиной 150 п. н. в КДС). Поиск ОРС был задан как специфичный для цепи, и единственное наилучшее совпадение было сохранено для дальнейшего анализа.
Все полные последовательности CDS были загружены в рабочий процесс функциональных аннотаций OmicsBox (эквивалентный более старой программе Blast2GO) (Gotz et al., 2008) со следующими измененными параметрами взрыва: plastp-fast, фильтр видов 89593 Craniata <хордовые>, охват HSP-Hit 70%. По умолчанию пороговое значение e-значения для шага blastp составляло 1,0E-3. Критерии охвата HSP-Hit гарантировали, что любые совпадения были похожи в большей части последовательности, а не просто содержали очень похожую частичную последовательность. Результаты взрывов были загружены в модули картографирования и аннотации GO. В полных CDS также проводили поиск функциональных мотивов с использованием InterProscan. Результаты были объединены в окончательный файл функциональной аннотации. Этот процесс позволил нам идентифицировать изоформы транскриптов мРНК с полными кодирующими областями, идентифицировать генные коды, где они не могли быть предоставлены SQANTI, и предоставить функциональное описание белков в каркасе GO, а также коды ферментов.
Последовательности, которые были картированы на митохондриальной последовательности Salmo salar с помощью SQANTI, также были переданы в TransDecoder с использованием генетического кода митохондрий позвоночных для поиска ORF. Любые полные CDS из этих последовательностей также были функционально аннотированы, как описано выше.
Кроме того, для транскриптов в окончательном полноразмерном транскриптоме мРНК (см. раздел «Окончательная фильтрация мРНК на основе подтверждающих доказательств и представление TSA») мы определили подгруппы полноразмерных мРНК, экспрессированных по крайней мере в трех образцах из определенного тип органа, но не в любом другом типе органов в наших материалах. Для них мы создали многоуровневые диаграммы GO, показывающие наиболее конкретные термины GO, появляющиеся в наборе данных, по сравнению с пороговыми значениями численности по умолчанию, предложенными OmicsBox, без избыточности. Эти отсечки составили соответственно 144 транскрипта для жабр, 98 в голове и почках и 116 в печени.
Окончательная фильтрация мРНК, основанная на подтверждающих доказательствах и представлении TSA
Для идентификации прочтений EC-HQ, представляющих транскрипты мРНК, использовался внутренний скрипт Python (Фильтр, рис. 3). Внутренний скрипт отфильтровывал все чтения EC-HQ, и сохранялись только чтения EC-HQ, которые TransDecoder прогнозировал как содержащие полный CDS. Кроме того, они должны быть классифицированы SQANTI как полное совпадение сплайсов, новые в каталоге или новые не в каталоге с включением канонических соединений сплайсов. Если SQANTI классифицировал по-разному или только сгруппировал Cogent, минимальная поддержка по крайней мере 5 прочтений FLNC использовалась в качестве порога для включения таких структурных изоформ в наш окончательный полноразмерный транскриптом мРНК. Таким образом, для изоформ с этими структурными классификациями использовались несколько более строгие критерии поддержки FLNC, поскольку они не имели такого же уровня поддержки в существующих аннотациях генома, как описанные выше FSM, NIC и NNC (для которых требовалось только минимальное 2 Поддержка FLNC должна быть классифицирована как штаб-квартира). Сценарий также собрал всю информацию о структурных и функциональных аннотациях для отфильтрованных расшифровок в файл tsv (дополнительный файл 1).
Необработанные данные секвенирования PacBio и Illumina были отправлены в базу данных NCBI SRA, а окончательные последовательности транскриптов в нашем полноразмерном транскриптоме мРНК были отправлены в Transcriptome Shotgun Assembly под номером GIYK00000000. Версия, описанная в этом документе, является первой версией, GIYK01000000.
Сравнение транскриптомов с применением анализа BLAST и аннотаций SQANTI3
Транскриптом мРНК 0496 de novo и мРНК Salmo salar в базе данных RefSeq. Полный набор транскриптов мРНК RefSeq Salmo salar был идентифицирован и загружен в виде файлов fasta с полными записями с использованием поиска нуклеотидов с фильтрацией в базе данных NCBI RefSeq. Их искали по нашему полноразмерному транскриптому мРНК с использованием blast 2.9.0 + с отсечкой e-value 1e-15 и outputfmt «6 std qcovhsp slen». Те же параметры поиска использовались при обратном сравнении с нашим транскриптомом в качестве запроса к последовательностям RefSeq. Для фильтрации результатов взрыва использовался внутренний скрипт Python. Фильтр классифицировал стенограммы по трем категориям. Совпадения между запросом и субъектом, отвечающие следующим критериям, были классифицированы как идентичные изоформы: Е-значение менее 10 –50 , процентная идентичность ≥ 99 % и либо покрытие запроса на пару сегментов с высокой оценкой > 99 %, либо длина выравнивания ∗ 100/длина субъекта > 99. Это гарантировало, что любое совпадение соответствует пороговым значениям e-значения и идентичности. имел более чем 99%-ное покрытие запроса последовательностью субъекта или более чем 99%-ное покрытие субъекта последовательности запроса. Эти пороги гарантировали, что совпадения, классифицированные как идентичные изоформы, были последовательно совпадающими последовательностями, происходящими из одних и тех же изоформ, но позволяли одной из них отличаться по длине UTR по сравнению с другой. Совпадения в этой категории были дополнительно сгруппированы в зависимости от того, имели ли РНК RefSeq более длинный UTR (покрытие запросов на пару сегментов с высокой оценкой < 99%) или имеет ли совпадающая мРНК в нашем полноразмерном транскриптоме мРНК более длинный UTR (длина выравнивания * 100/длина субъекта <99). Ко второй категории, названной значительными совпадениями, относились все совпадения, не соответствующие идентичным критериям изоформы, но со значением E менее 10 –15 . Остальные последовательности запросов, возвращающие E-значения более 10 –15 , были отнесены к категории несовпадающих мРНК RefSeq (или несовпадающих полноразмерных мРНК в обратном поиске).
Перекрытие между мРНК в нашем наборе данных и мРНК на основе геномной аннотации (эталонная последовательность генома GCF_000233375.1) будет числом последовательностей, классифицированных SQANTI3 как FSM. Они были получены из файла final.tsv (дополнительный файл 1). Для создания диаграммы Венна в разделе «Кластеризация и группировка Уникальные данные EC-HQ показали, что 22% из них не могут быть сопоставлены с текущей последовательностью генома атлантического лосося».
мРНК RefSeq с несовпадающими последовательностями по сравнению с коррелированными мРНК (экзонами) в сборке генома атлантического лосося идентифицировали путем добавления термина «И «разрыв сборки:» [Все поля]» к поиску в базе данных RefSeq. Они представляли РНК RefSeq, которые не поддерживаются текущей последовательностью генома. Кроме того, количество таких мРНК RefSeq, которые поддерживаются нашими полноразмерными мРНК, было получено путем поиска их инвентарных номеров среди тех, которые отнесены к идентичной категории изоформ, описанной выше.
Результаты
Гибридная коррекция ошибок повысила точность последовательности и позволила удалить артефакты секвенирования
Результаты секвенирования Pacbio и Illumina обобщены в дополнительном файле 2. Как и ожидалось, одна клетка Sequel II генерировала примерно 4–7 раз больше чтений HQ, чем две ячейки Sequel I, но процент чтений HQ, сгенерированных из чтений CCS, был одинаковым. Кроме того, распределение чтений по размерам с двух платформ показало очень похожее распределение (дополнительный файл 3), что указывает на то, что считывания, созданные двумя платформами, были одинакового качества. После фильтрации HQ было в общей сложности 2 080 166 чтений HQ, распределенных по 11 образцам (дополнительный файл 2). Количество прочтений Illumina во всех образцах, примененных для исправления ошибок, было более 9.00 миллионов со средним качеством phred 36. Это привело к 1 596 834 чтениям EC-HQ.
Распределение покрытия считываниями Illumina на считываниях HQ показано на рисунке 4, а точное количество чтений HQ, покрываемых определенным процентом чтений Illumina, указано в дополнительном файле 4. На рисунке показано, что большинство чтений HQ были сохранены (и исправлены ошибки) этим процессом фильтрации. Более 81% прочтений HQ имели охват 99% или более, что свидетельствует о том, что большинство прочтений HQ были с исправлением ошибок во всей их последовательности. Основной причиной удаления было не общее плохое освещение. Вместо этого последовательности, которые были удалены (23,3%), были сильно взвешены в сторону прочтений HQ с высоким покрытием (9охват 5–99%), но с небольшими внутренними пробелами в покрытии чтения Illumina. При 99% охвате было, например, приблизительно 100 000 прочтений HQ с внутренними пробелами, не охваченными графиком де Брюжина (DBG). Это может указывать на то, что эти чтения HQ были артефактами, созданными в конвейере PacBio, представляющими загрязняющие геномные последовательности или продукты слияния (разные транскрипты, слитые вместе и секвенированные SMRT). Это наиболее вероятное объяснение, поскольку только меньшие части последовательностей EC-HQ не были покрыты независимыми и гораздо более «глубокими» последовательностями транскриптома с платформы Illumina. Хотя степень коррекции одной п.н. не могла быть измерена напрямую, мы сравнили длины CDS до и после исправления ошибок. Это сравнение показало, что 118 199 (7%) прочтений увеличили размер ORF после коррекции, 1 455 732 (91%) имели ту же длину ORF, а 22 903 (2%) имели более короткую ORF. Таким образом, значительная часть прочтений была исправлена, и 75% из них увеличили длину своих CDS. Количество коротких чтений, примененных для исправления ошибок, составило более 927 миллионов (дополнительный файл 1). Это равно 20% всех коротких прочтений, используемых для аннотирования последовательностей экзома в текущей сборке генома.
Рис. 4. Распределение покрытия по LoRDEC для последовательностей с (оранжевые столбцы) и без (оранжевые столбцы) внутренними пропусками в интервале покрытия 75–100%.
Одна треть транскриптов представляла собой вкрапленные повторы
Набор данных чтения EC-HQ был проанализирован с помощью Repeatmasker для идентификации транскриптов, происходящих из вкрапленных повторов (Repeatmasker, рис. 2). После фильтрации любых прочтений EC-HQ, совпадающих с вкрапленными повторами, набор данных был сокращен до 1 090 532 прочтений EC-HQ. Это показало, что около трети (32%) всех транскриптов атлантического лосося представляют собой просто экспрессированные вкрапленные повторы.
Кластеризация и группировка уникальных прочтений EC-HQ показали, что 22% из них не могут быть сопоставлены с текущей последовательностью генома атлантического лосося
Картирование прочтений EC-HQ с геномом Salmo salar с помощью cDNA_cupcake (рис. 2) показало 89% успеха при картировании 972 904 прочтений EC-HQ. Они были сокращены до 87 315 уникальных чтений после кластеризации CD-Hit. Однако примерно половина (59 913 из 117 628) оставшихся прочтений EC-HQ может быть кДНК_капкейком, сгруппированным с геномом Salmo trutta . CD-Hit сократил их до 8721 уникального чтения. Остальные 57 715 чтений EC-HQ были сгруппированы с помощью Cogent, которые были сокращены до 16 367 уникальных чтений с помощью CD-Hit. cDNA_Cupcake и Cogent также присвоили каждой последовательности номер локуса. Таким образом, если они картировались в одном и том же месте генома или псевдогенома Cogent перекрывающимся образом, они были сгруппированы вместе как последовательности, которые, вероятно, были изоформами одного и того же транскрипта. В целом кластеризация cDNA_cupcake, Cogent и final CD-Hit сократила полный набор данных до 112 404 уникальных транскриптов EC-HQ (окончательный неизбыточный транскриптом EC-HQ, рисунок 2). Таким образом, 78% уникальных транскриптов EC-HQ были сопоставлены с Salmo salar геном, в то время как примерно 8% было сопоставлено с геномом Salmo trutta . Остальные 14% уникальных прочтений EC-HQ (кластеризованные Cogent) не могут быть сопоставлены ни с одним из двух геномов лососевых. Очень маловероятно, что восемь процентов транскриптов будут хорошо отображены в геноме Salmo trutta, если причиной несоответствия геному Salmo salar будет низкое качество или ошибка в считываниях EC-HQ. Напротив, это указывало на то, что в текущей последовательности генома атлантического лосося отсутствуют или неправильно собраны последовательности, которые препятствуют успешному картированию удивительно большой доли (22%) уникальных прочтений EC-HQ атлантического лосося. Исходя из этого, ошибки в последовательности генома могут быть вероятным объяснением того, почему 14% транскриптов, сгруппированных Cogent, не могут быть сопоставлены с 9Последовательность генома 0496 Salmo salar (или генома Salmo trutta ). Тот факт, что наш окончательный полноразмерный транскриптом намного лучше соответствует текущим мРНК RefSeq, чем транскрипты (аннотированные экзоны) в последовательности генома (идентичные изоформы против FSM, раздел «Сравнение полноразмерного транскриптома с аннотацией транскрипта генома с применением SQANTI3»). ») также указывает на неправильную аннотацию продуктов сращивания. Таким образом, полноразмерный транскриптом de novo из этого исследования может помочь улучшить текущую аннотацию транскрипта в последовательности генома. В свете этого 22% транскриптов, которые в настоящее время не сопоставлены с последовательностью генома атлантического лосося, представляют собой очень полезный источник информации о долгосрочных связях, которую можно использовать для улучшения сборки последовательности генома.
Транскриптом
de novo , состоящий из 71 461 полноразмерной мРНК из 23 071 локуса сопоставленные с геномом Salmo trutta , были структурно аннотированы с использованием SQANTI3 (рис. 3). Остальные последовательности, которые были сгруппированы Cogent, не могли быть осмысленно аннотированы таким образом, а скорее полагались на функциональные аннотации (OmicsBox, рис. 3) для классификации категорий РНК. Полное распределение структурных классификаций для всех неизбыточных чтений EC-HQ показано в таблице 2.
Таблица 2. Распределение SQANTI-классификаций и группировка Cogent для неизбыточного транскриптома высокого качества с исправлением ошибок (рис. 2) и отфильтрованного транскриптома мРНК (рис. 3).
Все 112 404 уникальных последовательности, независимо от метода кластеризации, также использовались для предсказания ORF с помощью Transdecoder и дальнейшей функциональной аннотации с помощью OmicsBox, если было предсказано, что они будут иметь полную CDS. После последнего этапа фильтрации (Фильтр, рис. 3) наш окончательный набор данных мРНК состоял из 71 461 неизбыточного чтения EC-HQ, которые с использованием SQANTI3 и наших критериев OmicsBox были аннотированы как транскрипты, кодирующие белок, с полной CDS. Было предсказано, что эти транскрипты происходят из 23 071 локуса или вероятных локусов в случае транскриптов Cogent, в среднем по три транскрипта на локус. Список прочтений EC-HQ, а также результаты анализа SQANTI, происхождение образца, поддержка FLNC, описания генов, коды GO, коды ферментов и идентификаторы контигов TSA представлены в дополнительном файле 1. Несмотря на то, что они классифицируются как антисмысловые, генные, межгенные или слияние SQANTI, эти категории содержали 2591, классифицированные TransDecoder как имеющие полную CDS и с поддержкой Pfam в качестве полноразмерных кодирующих белок мРНК. Дополнительные 4803 транскрипта, аналогичным образом классифицированные TransDecoder как мРНК, кодирующие полноразмерный белок, не были картированы ни с одним из геномов лососевых (кластеризовано Cogent). Шестьдесят семь процентов транскриптов, не сопоставленных или сопоставленных с категорией небелкового кодирования SQANTI3, также были подтверждены как транскрипты, кодирующие белок, с помощью следующей аннотации GO (раздел «Более 80% транскриптов были присвоены терминам и подмножествам GO»). Выявленные органоспецифические паттерны экспрессии») Таким образом, в общей сложности 71 461 уникальных транскриптов (63%) были классифицированы как мРНК, в то время как остальные транскрипты (37%), вероятно, представляли собой некодирующие РНК какого-то другого типа. Распределение мРНК по длине показано на рисунке 5. Длина транскриптов мРНК варьировалась от 319оснований до 13 331 основания в длину, со средней длиной 1402 п.н. и средней длиной 3209 п.н.
Рисунок 5. Окончательный полноразмерный транскриптом мРНК, распределенный по длине транскрипта. В каждом столбце показано количество транскриптов, попадающих в заданный интервал длины 500 п.н.
Чтения EC-HQ в окончательном наборе данных мРНК, отнесенном к категории Full Splice Match (FSM) (финал в таблице 2), представляют собой совпадение, идентичное известной изоформе в текущих аннотациях генома ( Salmo salar или Salmo trutta ) с точки зрения схемы сплайсинга и идентичности последовательности (SQANTI3 по умолчанию 95% отсечки). Большинство таких совпадающих транскриптов в текущей аннотации атлантического лосося были созданы на основе предсказаний, основанных на последовательности генома с переменной поддержкой коротких прочтений HTS или EST (96% мРНК Salmo salar RefSeq являются записями XM). Таким образом, FSM в нашем наборе данных представляют собой экспериментальную проверку 17 787 изоформ транскриптов с помощью полноразмерных мРНК, секвенированных одной молекулой. Удивительно, но их было 9.60 прочтений EC-HQ, картированных как FSM в геноме Salmo trutta. Очевидно, это также настоящие полноразмерные мРНК атлантического лосося, но несоответствия в текущей сборке генома Salmo salar помешали картированию этих транскриптов с последовательностью генома Salmo salar .
Транскрипты, обозначенные как «новый в каталоге» (NIC) или «новый не в каталоге» (NNC) с экзонами, определяемыми каноническими соединениями сплайсинга, представляют собой новые изоформы, которые в настоящее время не аннотированы в последовательностях генома. NIC имеют комбинации известных сайтов сплайсинга, что делает их изоформами с новыми комбинациями аннотированных экзонов. было 17 039таких новых транскриптов в окончательном наборе данных примерно столько же, сколько и во всех FSM. Было еще большее количество NNC, всего 25 581 транскрипт, что иллюстрирует способность методов, основанных на длинных чтениях, идентифицировать новые изоформы, которые невозможно надежно предсказать с использованием только коротких чтений и последовательности генома. Опять же, значительная часть (8,5%) транскриптов в этих категориях может быть картирована и классифицирована SQANTI3 только с использованием генома Salmo trutta .
Остальные категории SQANTI3 (ISM, антисмысловые, генные, межгенные и слитые) составляли меньшую часть конечного транскриптома мРНК (всего 5291 транскриптов). Несмотря на то, что их структурная классификация SQANTI3 ставит под сомнение, были ли это настоящие мРНК, все они были поддержаны OmicsBox как полноразмерные мРНК. Кроме того, поскольку для фильтрации возможных артефактов из этих категорий использовался порог не менее 5 поддерживающих FLNC, они, вероятно, представляют собой настоящие транскрипты, кодирующие белок атлантического лосося. Хотя считывания ISM подтверждались аннотацией генома как совпадающие с известным транскриптом, но с отсутствующей экзонной последовательностью на 5′- или 3′-конце (или на обоих), все они имели полную CDS. Мы использовали более консервативные критерии поддержки FLNC (не менее 5 FLNC) для ISM, и, учитывая этот порог, менее вероятно, что они являются неполными, а скорее представляют собой полноразмерные мРНК. Небольшое количество последовательностей ISM (3,7%) имело 80% или более содержания А в 20 основаниях непосредственно ниже того места, где они были нанесены на карту в последовательности генома. Это могло привести к неправильному праймированию во время синтеза кДНК, что привело к неправильной длине 3’UTR в этих транскриптах. Доля транскриптов, сгруппированных Cogent с не менее чем 5 поддерживающими FLNC, примерно такая же большая (4803 транскрипта), как и те, которые сопоставлены с Салмо трутта . Опять же, это иллюстрирует способность секвенирования транскриптома на основе длительного считывания идентифицировать транскрипты, не обнаруженные предсказаниями генома, поддерживаемыми коротким считыванием.
Обзор количества FLNC, поддерживающих каждую мРНК EC-HQ в окончательном полноразмерном транскриптоме мРНК, представлен на рисунке 6. На рисунке показано, что более 74% поддерживают более пяти FLNC независимо от категории. Это означает, что примерно 70% категорий FSM, NIC и NNC были поддержаны пятью или более FLNC, даже если критерием включения для этих категорий было два или более FLNC.
Рис. 6. Распределение полноразмерной неконкатемерной поддержки в окончательном наборе данных мРНК. В каждом столбце показано количество транскриптов в конечном транскриптоме мРНК с количеством полноразмерных неконкатемерных ридов, поддерживающих длинные риды.
Сценарий Python использовался для оценки количества случаев, когда транскрипты, аннотированные как кодирующие один и тот же ген SQANTI или OmicsBox, были назначены разным локусам cdna_Cupcake или разным семействам генов Cogent. В целом 12% генов имели по крайней мере два транскрипта мРНК EC-HQ, которые были картированы в разных локусах. Это указывает на то, что по крайней мере 12% экспрессированных генов, вероятно, были представлены несколькими паралогами в нашем наборе данных.
Стенограммы депонированы в DDBJ/EMBL/GenBank в качестве проекта Transcriptome Shotgun Assembly под регистрационным номером GIYK00000000. Версия, описанная в этом документе, является первой версией, GIYK01000000. Идентификатор TSA Contig ID для каждой последовательности указан в дополнительном файле 1. Сравнение
показало, что транскриптом
de novo лучше поддерживает транскрипты мРНК RefSeq, чем последовательность сборки генома Текущая информация о транскриптах последовательностей мРНК атлантического лосося предоставлена из два источника NCBI. Одним из них является информация о транскриптах (варианты транскриптов и изоформ, определяемые их экзонами), приведенная в аннотации к текущим 9Эталонная последовательность генома 0496 Salmo salar . Другим источником является текущая коллекция транскриптов мРНК Salmo salar в базе данных RefSeq. Хотя можно было бы ожидать, что они хорошо соответствуют друг другу, они различаются информацией о последовательностях, приведенной для тысяч транскриптов. Существует 4475 мРНК RefSeq, которые аннотированы как имеющие пробел или дополнительную последовательность, которой нет в текущей эталонной последовательности генома. Транскриптом мРНК de novo , основанный на длительном прочтении, из настоящего исследования, возможно, может помочь решить, какие последовательности транскриптов являются правильными. Ожидается, что применяемый здесь метод на основе одной молекулы будет лучшим для идентификации изоформ. Этот потенциал для повышения качества аннотированных изоформ транскриптов с помощью нашего набора данных был исследован путем сравнения нашего транскриптома длительного чтения с каждым из доступных источников (последовательности мРНК RefSeq и аннотация транскриптов в последовательности генома RefSeq).
Сравнение полноразмерного транскриптома и транскриптома RefSeq с применением BLASTN
мРНК Salmo salar в RefSeq состоят в основном из транскриптов, которые предсказаны с использованием последовательности генома, но поддерживаются и исправляются ошибки на основе EST и последовательностей с коротким считыванием, как часть конвейера аннотации генома эукариот NCBI 1 . Все 97 604 последовательностей мРНК Salmo salar в базе данных RefSeq были сопоставлены с нашим набором данных, классифицируя совпадения по трем категориям (рис. 7). Идентичные изоформы [99% идентичность и покрытие более короткой совпадающей последовательности более длинной совпадающей последовательностью (см. раздел «Сравнения транскриптомов с применением анализа BLAST и аннотаций SQANTI3» для более подробной информации)]. Второй категорией были значительные совпадения (все транскрипты со значениями e меньше 1e-15, но не отвечающие очень строгим критериям идентичности изоформ), а третьей категорией были названы несоответствующие мРНК RefSeq (все транскрипты со значениями e больше, чем 1e-15). чем Е-15 или без попаданий). При фильтрации в соответствии с этими критериями 24 415 (25%) транскриптов RefSeq относились к категории идентичных изоформ, что обеспечивает экспериментальную проверку четверти изоформ в базе данных RefSeq с помощью полноразмерных мРНК из нашего транскриптома с длительным чтением. Кроме того, было примерно в два раза больше мРНК со значительными совпадениями (49).785) против нашего транскриптома, что указывает на то, что дополнительная половина всех транскриптов RefSeq присутствовала в нашем наборе данных в виде сплайс-вариантов или паралогов. Учитывая, что части последовательностей мРНК RefSeq предсказаны из последовательности генома (96% составляют записи XM), существует также вероятность того, что некоторые совпадения в категории значимых совпадений на самом деле являются идентичными изоформами, но не соответствуют самому строгие критерии, которые мы применили для этой категории из-за ошибок последовательности. 23 404 мРНК в категории несовпадающих RefSeq-мРНК, вероятно, представляют собой транскрипты генов, не экспрессирующихся в органах, включенных в это исследование. мРНК в категории идентичных изоформ показали распределение различий в длине, при этом более длинный транскрипт был взят из набора данных RefSeq в 42% случаев, в то время как два совпадающих транскрипта отклонялись менее чем на одну сотую своей длины в 28% случаев, а в остальные 30% более длинных расшифровок были из наших de novo транскриптом. Эти различия в размерах в большинстве случаев были небольшими и затрагивали только UTR, а не CDS.
Рисунок 7. Круговая диаграмма , показывающая распределение результатов blast при поиске всех мРНК Salmo salar RefSeq по сравнению с окончательным набором данных мРНК полной длины. Синие — идентичные изоформы, оранжевые — значимые совпадения, серые — несовпадающие мРНК RefSeq.
Мы также провели обратное сравнение с новым поиском blastn, где нашим набором данных были последовательности запросов против Salmo salar RefSeq мРНК (рис. 8). Это показало, насколько хорошо наш транскриптом представлен в текущем транскриптоме RefSeq. Примечательно, что количество последовательностей, по крайней мере с одним бластным попаданием, отвечающим идентичным критериям изоформ, было ниже при использовании полноразмерного транскриптома в качестве запроса, чем при использовании мРНК RefSeq в качестве запроса (20 582, рис. 8 против 24 415, рис. 7). Это указывает на то, что некоторые из наших транскриптов были классифицированы как идентичные изоформам нескольких последовательностей, которые в настоящее время перечислены отдельно в RefSeq (рис. 7). Некоторые возможные объяснения этого открытия заключаются в том, что некоторые последовательности в RefSeq являются избыточными и/или что некоторые из наших расшифровок имеют неполные UTR, из-за чего они не могут различать некоторые записи RefSeq. Сравнение также показало, что только 566 наших последовательностей не имели значительного попадания взрыва (<1e-15). Таким образом, результат обратного бластн-анализа показал, что наш транскриптом, имеющий 99% поддержки в RefSeq состояли почти исключительно из вариантов транскриптов известных генов, а не из транскриптов новых генов (рис. 8). Взятые вместе, два бластных анализа показали, что наш полноразмерный транскриптом de novo подтвердил 25% известных в настоящее время транскриптов атлантического лосося в RefSeq, предоставил большое количество новых изоформ, значительно совпадающих с 50% известных транскриптов в RefSeq, но не обнаружили много транскриптов новых генов (1%, рис. 8).
Рис. 8. Круговая диаграмма , показывающая распределение результатов бластов при поиске в окончательном наборе данных полной длины мРНК по всем мРНК Salmo salar RefSeq. Синие — идентичные изоформы, оранжевые — значительные совпадения, серые — несовпадающие новые мРНК.
Сравнение полноразмерного транскриптома с аннотацией транскрипта генома с применением SQANTI3
На рис. 9 показано распределение транскриптов общих изоформ при сравнении нашего полноразмерного транскриптома с мРНК в Salmo salar аннотация генома. На рисунке показано, что 17 782 транскрипта мРНК EC-HQ полностью соответствовали (идентичны) сплайсингу уже аннотированным транскриптам в версии RefSeq сборки генома Salmo salar . Большинство изоформ мРНК, предсказанных в аннотации генома Salmo salar (81%, мРНК Salmo salar без FSM на рисунке 9), не могут быть подтверждены нашими транскриптами мРНК с длительным чтением. Кроме того, в нашем окончательном наборе данных было 53 674 мРНК (75%, новые мРНК на рис. 9).), которые представляли собой новые изоформы, картированные в геноме (61%, категории, не относящиеся к FSM, в Salmo salar в таблице 2), или мРНК, которые вообще не картировались (14%, категории Salmo trutta или Cogent в таблице 2). ) из-за несоответствия на уровне последовательности генома.
Рисунок 9. Диаграмма Венна, иллюстрирующая количество идентичных изоформ (Full Slice-Match), общих для мРНК в аннотации генома и конечного полноразмерного транскриптома мРНК. Ни один FSM не представляет изоформы в аннотации генома без идентичного совпадения в конечном полноразмерном транскриптоме мРНК. Новые мРНК относятся к изоформам транскриптов в окончательном наборе данных мРНК без идентичного совпадения с последовательностями, аннотированными в 9Сборка генома 0496 Salmo Salar .
Тот факт, что FSM было значительно меньше (19%, рис. 9), чем количество идентичных изоформ (25%, рис. 7), может указывать на то, что последовательность генома является менее надежным источником последовательностей транскриптов. Это также подтверждается тем фактом, что 14% транскриптов мРНК вообще не картировались (сопоставлены с Salmo trutta или сгруппированы Cogent). Кроме того, 1268 транскриптов, которые не картировались с геномом Salmo salar , относились к категории идентичных изоформ при сравнении бластов с мРНК RefSeq. Аналогичное сравнение, в котором транскрипты RefSeq, не соответствующие эталонной последовательности генома (4475 транскриптов, методы, «Сравнения транскриптомов с применением анализа BLAST и аннотаций SQANTI3»), сравнивались с нашими de novo транскриптом показал, что 673 из них относятся к категории идентичных изоформ. Все вместе эти сравнения показывают, что полноразмерный транскриптом de novo может внести значительный вклад в улучшение качества аннотаций текущего транскрипта и помочь в улучшении сборки эталонного генома.
Более 80% расшифровок были присвоены GO Термины и подмножества Выявленные органоспецифические паттерны экспрессии
На рис. 10 показано распределение результатов аннотации OmicsBox в окончательном наборе данных мРНК. Восемьдесят два процента транскриптов были успешно аннотированы по крайней мере одним термином GO (GOs> 0, рисунок 10). Остальные 18% транскриптов без терминов GO были распределены на транскрипты со значительными бласт-хитами, но на белки без терминов GO в базе данных Gene Ontology (9).%), в то время как у другой половины не было значительных совпадений в базе данных белков RefSeq. Вместо этого они были подтверждены как кодирующие белок их длиной CDS и поддержкой в Pfam. Функциональная аннотация, включая термины GO и символы генов, включена в дополнительный файл 1. Распределение количества терминов GO, присвоенных каждой последовательности, показало, что 50% транскриптов в наборе данных были назначены между 2 и 5 терминами GO, в то время как 14% были назначены еще больше. Вместе мы получили солидный уровень функциональной аннотации примерно для двух третей нашего набора данных. Всего 46 769из них также были аннотированы конкретными генными символами на основе результатов Blastp от OmicsBox.
Рисунок 10. Распределение числа предсказанных терминов Генной онтологии. В каждом столбце показано количество расшифровок, попадающих в заданный интервал терминов онтологии генов, определенных для расшифровки.
Десять процентов всех FLNC в наборе данных были получены только из 17 мРНК (перечислены в дополнительном файле 5). Это продемонстрировало, что в конечном транскриптоме мРНК было несколько высокоэкспрессированных транскриптов, кодирующих белок. Единственная самая многочисленная стенограмма, alb1 , составляли 3,8% всех FLNC сами по себе, а первые пять наиболее распространенных транскриптов составляли 6,5% всех FLNC. Сывороточный альбумин ( alb1 ) и два других наиболее экспрессируемых гена ( fgg и itih4 ) кодировали секреторные белки из тканей печени. Другие гены с высокой экспрессией, такие как два варианта сплайсинга актина ( actb ), селенопротеин P ( SelP ) и аполипопротеин Eb ( apoeb ), экспрессировались во всех тканях. Удивительным открытием стало то, что один из высокоэкспрессированных транскриптов, аннотированный как H-подобный фактор комплемента ( cfhr5 ) не соответствовал последовательности генома атлантического лосося, но был FSM в геноме Salmo trutta . Кроме того, два других транскрипта с высокой экспрессией (протромбин-подобный и трансферрин-А-подобный) были аннотированы SQANTI как продукты слияния. Опять же, это, вероятно, связано с неправильной аннотацией генома, а не с артефактами последовательности, учитывая большое количество FLNC, поддерживающих эти транскрипты в нескольких тканях.
Окончательные транскрипты мРНК были получены из трех органов: печени, жабр и головного мозга. Некоторые из транскриптов с высокой экспрессией присутствовали только в образцах печени, что указывает на то, что конвейер транскриптома может идентифицировать транскрипты, экспрессируемые органоспецифическим образом. Применяя консервативный подход к идентификации таких органоспецифических транскриптов, мы искали в полноразмерном транскриптоме мРНК транскрипты, которые были экспрессированы по крайней мере в трех образцах из одного органа, в то время как они отсутствовали в образцах из любого из двух других органов. Это показало, что 2717 транскриптов экспрессируются только в жабрах, либо из генов, экспрессируемых только в жабрах (1811), либо сплайс-вариантов, экспрессируемых только в жабрах (9).06). В печени было 1784 транскрипта, из которых 1113 были из генов, специфичных для печени, и 671 были специфичными для печени сплайс-вариантами. В головной почке было 1757 транскриптов, 700 из генов, специфичных для головной почки, и 1057 были специфичными для головной почки сплайс-вариантами. На рисунках 11–13 показано распределение наиболее специфических общих терминов Biological Process GO (см. Материалы и методы, «Функциональная аннотация») для этих трех органоспецифических групп мРНК. Каждый термин GO указывает на биологические процессы, которые обогащены транскриптами, специфичными для органа. GO-термины в транскриптах жабр ясно указывают на специфические функции жабр, такие как транспорт ионов и сигнальный путь рецептора клеточной поверхности, гены, которые принимают участие в осморегуляции. Транскрипты, аннотированные как играющие роль в развитии системы (например, развитие конкретных типов тканей и регуляция транскрипции ДНК), также специфически экспрессировались в образцах жабр (рис. 11). Головная почка у костистых рыб состоит из нескольких тканей с различными функциями, такими как экскреция, биосинтез стероидов и иммунный ответ. Среди транскриптов, специфически экспрессируемых в головной почке, были транскрипты, участвующие в биосинтезе макромолекул, ароматических и азотистых соединений. Многие также были отмечены как участвующие в организации и транспорте органелл (рис. 12). Было 132 иммунных функции и иммунного ответа генов, экспрессируемых только в голове и почке, хотя они не были признаны единой группой иммунных генов с помощью применяемых здесь методов GO (наиболее конкретные общие термины GO). Некоторые примеры таких транскриптов включают VIG2, различные INF, хемокины и толл-подобные рецепторы. Транскрипты в печени (рис. 13) показали термины GO, связанные с такими процессами, как метаболизм, биосинтез и свертывание крови (например, высоко экспрессированный fgg и itih4 ). Опять же, транскрипты, экспрессируемые исключительно в этом органе, как и ожидалось, оказались среди транскриптов, кодирующих белки, связанные с функцией печени. Взятые вместе, результаты здесь показали потенциал нашего конвейера транскриптома для идентификации генов и вариантов сплайсинга, которые имеют определенные органоспецифические функции.
Рис. 11. Диаграмма многоуровневой онтологии генов, Джилл. На круговой диаграмме показаны наиболее специфические термины генной онтологии, встречающиеся по крайней мере в 144 транскриптах, специфичных для жабр, без избыточности (см. также раздел «Функциональная аннотация» в разделе «Материалы и методы»).
Рисунок 12. Диаграмма Multilevel Gene Ontology, голова-почка. На круговой диаграмме показаны наиболее специфические термины генной онтологии, встречающиеся по крайней мере в 98 транскриптах, специфичных для головы и почек, без избыточности (см. также «Функциональная аннотация» в разделе «Материалы и методы»).
Рис. 13. Диаграмма Multilevel Gene Ontology, печень. На круговой диаграмме показаны наиболее специфические термины генной онтологии, встречающиеся по крайней мере в 116 транскриптах, специфичных для жабр, без избыточности (см. также Материалы и методы «Функциональная аннотация»).
Обсуждение
Преимущества применения гибридной коррекции ошибок одномолекулярных длинных чтений при секвенировании транскриптома
Наш проект был направлен на создание полноразмерного транскриптома мРНК с точностью последовательности референсного уровня из различных образцов органов путем обработки PacBio long-reads. считывания с гибридным исправлением ошибок с считываниями парных концов Illumina из тех же образцов. Подобные подходы использовались для создания транскриптомов высокого качества у других видов (Feng et al., 2019).; Puglia et al., 2020), но это первое в своем роде исследование атлантического лосося. Стратегия и основные функции конвейера показаны на рисунках 1–3. Во-первых, первоначальное создание консенсусных последовательностей из отдельных молекул, удаление артефактов и консервативная кластеризация были достигнуты с помощью пакета SMRTLink IsoSeq3 (PacBio, 2020; рисунок 1). Эта обработка исправляет большую часть высокой частоты необработанных ошибок, связанной с секвенированием PacBio [по оценкам, 11–14% (Roberts et al., 2013)]. Результатом являются последовательности (называемые считываниями HQ), которые поддерживаются как минимум двумя одиночными секвенированными молекулами с прогнозируемой точностью не менее 99%.
Первоначальная проверка прочтений HQ в материалах показала, что было много случаев ошибок сдвига рамки, приводящих к неправильным CDS или преждевременным стоп-кодонам (данные не показаны). Хотя мы не могли с уверенностью заключить, что во всех случаях это были ошибки секвенирования, такие ошибки не были неожиданными, учитывая точность последовательности 99% и тот факт, что считывания PacBio подвержены числовым ошибкам в гомополимерах (Tedersoo et al., 2018). Этой точности было бы недостаточно для нашей цели, поскольку мы стремились создать последовательности транскриптов с качеством столь же высоким (или лучше, чем) мРНК атлантического лосося в RefSeq. Применение подхода, при котором длинные чтения исправлялись с помощью данных секвенирования коротких чтений с последующей фильтрацией последовательностей, не поддерживаемых обоими наборами данных, казалось лучшим решением (Au et al., 2012). Подход с исправлением ошибок использует превосходную производительность платформы Pac Bio для идентификации структурных изоформ (и различий между очень похожими паралогами) путем секвенирования одной молекулы с длительным считыванием (Liang et al., 2016), в то время как точность, как ожидается, будет значительно выше. увеличивается из-за гораздо более высокой глубины чтения и качества phred, обеспечиваемого более короткими чтениями. Кроме того, тип ошибок, наиболее часто получаемых на двух платформах, неодинаков, и вероятность получения одной и той же загрязняющей последовательности из геномной ДНК или других артефактов слияния при обработке одного и того же образца в двух независимых методах синтеза кДНК и разных методах подготовки библиотеки маленький. Вместе последовательности транскриптов, созданные с помощью отдельных методов обработки образцов РНК, вероятно, снижали вероятность сохранения последовательностей с ошибками, генерируемыми в каждом из двух конвейеров обработки образцов, в окончательных отфильтрованных считываниях EC-HQ (рис. 2).
Предыдущее исследование (Sahraeian et al., 2017), в котором сравнивались различные инструменты для анализа последовательностей РНК, определило LoRDEC (Salmela and Rivals, 2014) как эффективный и точный инструмент для гибридной коррекции ошибок, и LoRDEC был успешно реализован в нашем трубопровод. Мы применили фильтрацию чтений EC-HQ, которая гарантировала удаление длинных чтений с внутренними пробелами, не поддерживаемыми более короткими чтениями. Вместе подход к исправлению ошибок с очень надежной поддержкой коротких чтений и дополнительным нижним порогом для поддержки FLNC гарантировал, что окончательные de novo последовательности транскриптома имели, в соответствии с результатами аналогичных исследований (Au et al., 2012), точность, сравнимую с другими справочными источниками транскриптов атлантического лосося.
Этот проект направлен на определение характеристик РНК, кодирующих белки. Поэтому в нашем конвейере был реализован программный пакет RepeatMasker, чтобы гарантировать, что окончательные последовательности не будут содержать транскрипты из длинных перемежающихся повторов. Треть последовательностей была идентифицирована как своего рода длинные вкрапленные повторяющиеся транскрипты. Это была удивительно большая доля всех расшифровок. Будущие проекты по транскриптомам атлантического лосося получат большую пользу от удаления таких транскриптов до синтеза кДНК и подготовки библиотеки (Жулидов, 2004). Кроме того, после фильтрации с помощью RepeatMasker значительная часть (37%) уникальных прочтений EC-HQ все еще не была классифицирована как транскрипты, кодирующие белок. Это показало, что наш конвейер, вероятно, идентифицировал тысячи длинных некодирующих РНК (днРНК). Характеристика днРНК с помощью подходов длительного считывания стала золотым стандартом для изучения днРНК (Wan et al., 2019).), и характеристика lncRNAs атлантического лосося из этих материалов в настоящее время продолжается в рамках параллельного проекта (рукопись готовится).
Полноразмерный транскриптом мРНК значительно увеличил количество изоформ
Независимо от того, сравниваете ли мы наши данные с аннотацией последовательности генома (SQANTI3) или анализом бластов с мРНК RefSeq атлантического лосося, около 70% окончательного транскриптома мРНК представляют собой новые изоформы. Эти новые изоформы представляют собой либо варианты сплайсинга (каждый локус имел в среднем три варианта сплайсинга), либо паралоги. Это показало, что наш подход к секвенированию длинного считывания транскриптома привел к существенному увеличению числа изоформ транскриптов атлантического лосося. Такой высокий уровень успеха в открытии новых изоформ хорошо согласуется с результатами аналогичных исследований (Zhang et al., 2019).). Для фильтрации оставшихся категорий транскриптов было реализовано отсечение из 5 поддерживающих FLNC. Хотя стандартное ограничение, рекомендованное разработчиком, составляет 10 FLNC (Tseng, 2020a), в других недавних исследованиях утверждается, что 5 FLNC достаточно для поддержки таких категорий, как транскрипты слияния (Nattestad et al., 2018). Мы пришли к выводу, что этап гибридной коррекции ошибок с минимум тремя поддерживающими чтениями Illumina по всей последовательности предоставил дополнительные подтверждающие доказательства, необходимые для принятия оставшихся категорий SQANTI и расшифровок Cogent при поддержке 5 FLNC. Хотя они были классифицированы SQANTI3 как сомнительные транскрипты мРНК или вообще не картированы, мы считаем вероятным, что это также настоящие полноразмерные мРНК, но они неправильно аннотированы в текущей последовательности генома.
Длинночитаемый транскриптом как ссылка на исследование экспрессии вариантов сплайсинга и паралогов из органов или особых условий Чтения FLNC были из 17 наиболее распространенных транскриптов). В будущих проектах, направленных на характеристику всех полноразмерных мРНК в образце материала, удаление таких транскриптов (а также всех вкрапленных повторов) значительно увеличит вероятность идентификации более редко экспрессируемых транскриптов (Жулидов, 2004).
Глубина секвенирования с использованием одной клетки Sequel II была примерно в восемь раз выше, чем с использованием одной клетки Sequel I. Это согласуется с другими исследованиями (Castaño et al., 2020; Lang et al., 2020). Сочетая высокую глубину чтения из Sequel II с методами нормализации для удаления обильных транскриптов, можно было бы ожидать, что большинство транскриптов, экспрессируемых в образце, будут обнаружены. Результаты этого исследования продемонстрировали, что наш конвейер имел возможность идентифицировать большое количество транскриптов, однозначно выраженных в каждом из трех типов органов, включенных в материалы. Следующая функциональная аннотация также показала, что наиболее распространенные термины GO, аннотированные с транскриптами, в значительной степени соответствовали функции этих органов. Кроме того, любые материалы, исследованные с помощью этого подхода с длительным чтением, могут идентифицировать не только уникально экспрессированные гены, но и уникально экспрессированные сплайс-варианты. Это также было продемонстрировано в группе транскриптов, однозначно экспрессируемых в одном органе.
Полноразмерные транскриптомы имеют ряд полезных применений (Oikonomopoulos et al., 2020). Среди упомянутых мы предполагаем, что наши высококачественные полноразмерные транскриптомы могут служить ссылками в анализе экспрессии. Последовательность генома атлантического лосося может быть менее подходящей в качестве такого эталона, поскольку очень большая часть транскриптов в этом исследовании не выровнена должным образом. Различие между вариантами сплайсинга или паралогами путем сопоставления коротких прочтений с последовательностью генома было бы подвержено ошибкам (если не невозможно). Вместо этого транскриптом длительного считывания с исправлением ошибок, представляющий уникальные хорошо охарактеризованные варианты транскриптов, может применяться в качестве эталона для определения того, какие последовательности транскриптов с платформ для секвенирования с коротким считыванием, обеспечивающие большую глубину считывания при доступных затратах, можно выровнять, подсчитать и проанализировать. с помощью таких инструментов, как DESeq2 (Love et al., 2014). Дополнительным преимуществом такого анализа является то, что они одновременно обнаруживают вариации SNP. UTR являются богатым источником таких вариаций (Andreassen et al., 2010), и такое картирование потенциально может выявить аллель-специфическую транскрипцию, быть примененным для обнаружения QTL и даже выявить причинную изменчивость, приводящую к фенотипическим различиям в сравниваемых группах.
В заключение следует отметить, что использованный здесь конвейер гибридного скорректированного длинного считывания успешно генерировал высококачественные полноразмерные транскрипты мРНК. Подход с длинным считыванием привел к обнаружению новых вариантов сплайсинга и подтвердил четверть всех предсказанных мРНК атлантического лосося с помощью транскриптов, происходящих из длинных чтений, секвенированных одной молекулой. Состоящие исключительно из мРНК с полными CDS, более 80% были присвоены термины GO, и были идентифицированы тысячи генов или вариантов сплайсинга генов, экспрессируемых органоспецифическим образом. Этот полноразмерный транскриптом станет важным ресурсом для функциональной геномики в исследованиях аквакультуры лосося.
Заявление о доступности данных
Наборы данных, представленные в этом исследовании, можно найти в онлайн-репозиториях. Названия репозитория/репозиториев и инвентарные номера можно найти в статье/дополнительных материалах.
Заявление об этике
Исследование на животных было рассмотрено и одобрено Национальным исследовательским органом Норвегии (NARA).
Вклад авторов
SR, RA, BH и T-KØ внесли свой вклад в концептуализацию и окончательное редактирование рукописи. SR и RA внесли свой вклад в разработку аналитического пайплайна, формальный анализ, а также написали, рассмотрели и отредактировали рукопись. SR участвовал в реализации конвейера, дополнительном программировании и написании исходного проекта. RA, BH и T-KØ внесли свой вклад в ресурсы. RA руководил и получал финансирование. RA и BH внесли свой вклад в администрирование проекта. Все авторы внесли свой вклад в статью и одобрили представленную версию.
Финансирование
Это исследование финансировалось грантом Норвежского исследовательского совета для RA (280839/E40).
Конфликт интересов
Авторы заявляют, что исследование проводилось при отсутствии каких-либо коммерческих или финансовых отношений, которые могли бы быть истолкованы как потенциальный конфликт интересов.
Благодарности
Мы хотели бы поблагодарить Саймона Вели из Норвежского ветеринарного института за предоставление контрольной ткани SAV и контрольных тканей, использованных в этом исследовании. Мы хотели бы поблагодарить Тину Висновску из Центра биоинформатики Университета Осло за помощь в устранении неполадок, техническую помощь и советы при разработке конвейера. Мы также благодарны Элизабет Ценг из Pacific Biosciences за помощь и плодотворные обсуждения во время разработки нашего пайплайна анализа, а также за разрешение использовать рисунок 1.9. https://www.ncbi.nlm.nih.gov/genome/annotation_euk/process/
Ссылки
Abdelrahman, H., ElHady, M., Alcivar-Warren, A., Allen, S., Al-Tobasei, R., Bao, L., et al. (2017). Геномика аквакультуры, генетика и селекция в Соединенных Штатах: текущее состояние, проблемы и приоритеты для будущих исследований. BMC Genomics 18:191. doi: 10.1186/s12864-017-3557-1
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
Аджубей А. А., Власова А. В., Хаген-Ларсен Х., Руден Т. А., Лаэрдал Дж. К. и Хойхайм Б. (2007). Аннотированные теги экспрессированных последовательностей (EST) из атлантического лосося перед смолтом ( Salmo salar ) в доступном для поиска ресурсе данных. BMC Genomics 8:209. doi: 10.1186/1471-2164-8-209
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
Аллендорф, Ф. В., и Торгаард, Г. Х. (1984). «Тетраплоидия и эволюция лососевых рыб», в Evolutionary Genetics of Fishes. Монографии по эволюционной биологии, изд. Би Джей Тернер (Бостон, Массачусетс: Springer), 1–53. doi: 10.1007/978-1-4684-4652-4_1
CrossRef Полный текст | Академия Google
Андреассен Р., Луннер С. и Хойхейм Б. (2009). Характеристика вставок полноразмерной секвенированной кДНК (FLIcs) из атлантического лосося ( Salmo salar ). BMC Genomics 10:502. doi: 10.1186/1471-2164-10-502
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
Андреассен Р., Луннер С. и Хойхайм Б. (2010). Направленное обнаружение SNP в генах атлантического лосося ( Salmo salar ) с использованием подхода обнаружения SNP с 3’UTR-примированием. BMC Геномика 11:706. doi: 10.1186/1471-2164-11-706
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
Андреассен Р. , Волдемариам Н. Т., Эгеланд И. О., Агафонов О., Синдре Х. и Хойхейм Б. (2017). Идентификация дифференциально экспрессируемых микроРНК атлантического лосося, реагирующих на инфекцию альфавирусом лосося (SAV). BMC Genomics 18:349. doi: 10.1186/s12864-017-3741-3
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
Эндрюс, С. (2010). FastQC : Инструмент контроля качества для высокопроизводительных данных о последовательностях [онлайн]. Доступно в Интернете по адресу: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (по состоянию на 2020 г.)
Google Scholar
Ау, К. Ф., Андервуд, Дж. Г., Ли, Л., и Вонг, У. Х. (2012). Улучшение точности длинных чтений PacBio за счет выравнивания коротких чтений. PLoS One 7:e46679. doi: 10.1371/journal.pone.0046679
PubMed Abstract | Полный текст перекрестной ссылки | Академия Google
Бернхардт, Л. В., Мирмель, М., Лиллехауг, А., Квиллер, Л., и Чиома Вели, С. (2021). Фильтрация, концентрация и обнаружение альфавируса лосося в морской воде во время совместного заражения лосося после смолта ( Salmo salar ). Дис. Аква. Орг. 144, 61–73. doi: 10.3354/dao03572
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
BioBam (2019). OmicsBox — биоинформатика стала проще [онлайн]. Валенсия: биоинформатика BioBam
Google Scholar
Бьорген Х. и Коппанг Э. О. (2021). Анатомия иммунных структур и органов костистых рыб. Иммуногенетика 73, 53–63. doi: 10.1007/s00251-020-01196-0
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
Кэмпбелл, М. А., Хейл, М. К., МакКинни, Г. Дж., Николс, К. М., и Пирс, Д. Э. (2019). Долгосрочное сохранение онологов за счет частичной тетрасомии после дупликации всего генома у лососевых. G3 (Бетесда) 9, 2017–2028 гг. doi: 10.1534/g3.119.400070
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
Castaño, C. , Berlin, A., Brandström Durling, M., Ihrmark, K., Lindahl, B.D., Stenlid, J., et al. (2020). Оптимизированное метабаркодирование с Pacific biosciences позволяет проводить полуколичественный анализ грибковых сообществ. Новый Фитол. 228, 1149–1158. doi: 10.1111/nph.16731
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
Конеса А., Мадригал П., Тарасона С., Гомес-Кабреро Д., Сервера А., Макферсон А. и др. (2016). Обзор лучших практик анализа данных секвенирования РНК. Геном Биол. 17:13. doi: 10.1186/s13059-016-0881-8
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
ФАО (2018). Состояние мирового рыболовства и аквакультуры, 2018 г. Рим: Продовольственная и сельскохозяйственная организация Объединенных Наций.
Google Scholar
Фэн С., Сюй М., Лю Ф., Цуй К. и Чжоу Б. (2019). Реконструкция полноразмерного атласа транскриптома с использованием PacBio Iso-seq дает представление об альтернативном сплайсинге в Gossypium australe . BMC Растение Биол. 19:365. doi: 10.1186/s12870-019-1968-7
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
Фу Л., Ниу Б., Чжу З., Ву С. и Ли В. (2012). CD-HIT: ускорено для кластеризации данных секвенирования нового поколения. Биоинформатика 28, 3150–3152. doi: 10.1093/bioinformatics/bts565
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
Джуффра Э., Таггл С.К. и Консорциум Ф. (2019 г.). Функциональная аннотация геномов животных (FAANG): текущие достижения и дорожная карта. год. Преподобный Аним. Бионауч. 7, 65–88. doi: 10.1146/annurev-animal-020518-114913
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
Гордон С. П., Ценг Э., Саламов А., Чжан Дж., Мэн X., Чжао З. и др. (2015). Широко распространенные полицистронные транскрипты у грибов, обнаруженные с помощью секвенирования одиночных молекул мРНК. PLoS One 10:e0132628. doi: 10.1371/journal. pone.0132628
Резюме PubMed | Полный текст перекрестной ссылки | Google Scholar
Gotz, S., Garcia-Gomez, J.M., Terol, J., Williams, T.D., Nagaraj, S.H., Nueda, M.J., et al. (2008). Высокопроизводительные функциональные аннотации и интеллектуальный анализ данных с пакетом Blast2GO. Рез. нуклеиновых кислот. 36, 3420–3435. doi: 10.1093/nar/gkn176
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
Хаас Б. и Папаниколау А. (2015). TransDecoder 5.5.0 [Онлайн]. Доступно в Интернете по адресу: https://github.com/TransDecoder/TransDecoder/wiki (по состоянию на 2019 г.).)
Google Scholar
Haas, B.J., Papanicolaou, A., Yassour, M., Grabherr, M., Blood, P.D., Bowden, J., et al. (2013). Реконструкция последовательности транскрипта de novo из РНК-seq с использованием платформы trinity для эталонного создания и анализа. Нац. протокол 8, 1494–1512. doi: 10.1038/nprot.2013.084
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
Хаген-Ларсен Х. , Лэрдал Дж. К., Паниц Ф., Аджубей А. и Хойхейм Б. (2005). Подход, основанный на EST, для идентификации генов, экспрессируемых в кишечнике и жабрах атлантического лосося перед смолтом (9).0496 Салмо Салар ). BMC Genomics 6:171. doi: 10.1186/1471-2164-6-171
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
Хьельтнес Б., Банг-Йенсен Б., Борнё Г., Хаукаас А. и Вальде К. С. (редакторы) (2019). Состояние здоровья в аквакультуре Норвегии, 2018 г. Осло: Норвежский ветеринарный институт.
Google Scholar
Hoar, WS (1988). 4 физиология неющих лососевых рыб. Физиол. Дев. Рыба живородящая молодь. 11, 275–343. doi: 10.1016/s1546-5098(08)60216-2
CrossRef Full Text | Google Scholar
Хьюстон, Р. Д., и Маккуин, Д. Дж. (2019). Генетика атлантического лосося ( Salmo salarL. ) в 21 веке: скачки вперед в аквакультуре и понимании биологии. Аним. Жене. 50, 3–14. doi: 10.1111/age.12748
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
Хабли Р. , Финн Р. Д., Клементс Дж., Эдди С. Р., Джонс Т. А., Бао В. и др. (2016). База данных Dfam семейств повторяющихся ДНК. Рез. нуклеиновых кислот. 44, Д81–Д89. doi: 10.1093/nar/gkv1272
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
Куп, Б. Ф., фон Шальбург, К. Р., Леонг, Дж., Уокер, Н., Лиеф, Р., Купер, Г. А., и др. (2008). Геномное исследование EST лососевых: гены, дупликации, филогения и микрочипы. BMC Genomics 9:545. doi: 10.1186/1471-2164-9-545
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
Ланг Д., Чжан С., Рен П., Лян Ф., Сунь З., Мэн Г. и др. (2020). Сравнение двух современных технологий секвенирования для сборки генома: чтения HiFi системы Pacific Biosciences Sequel II и сверхдлинные чтения Oxford Nanopore. GigaScience 9:giaa123. doi: 10.1093/gigascience/giaa123
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
Леонг Дж. С., Янцен С. Г., фон Шальбург К. Р., Купер Г. А., Мессмер А. М., Ляо Н. Ю. и др. (2010). Полноразмерные последовательности кДНК Salmo salar и Esox lucius показывают изменения в эволюционном давлении на геном после тетраплоидизации. BMC Genomics 11:279. doi: 10.1186/1471-2164-11-279
PubMed Abstract | Полный текст перекрестной ссылки | Академия Google
Ли, Х., и Бироль, И. (2018). Minimap2: попарное выравнивание нуклеотидных последовательностей. Биоинформатика 34, 3094–3100. doi: 10.1093/bioinformatics/bty191
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
Ли В. и Годзик А. (2006). Cd-hit: быстрая программа для кластеризации и сравнения больших наборов последовательностей белков или нуклеотидов. Биоинформатика 22, 1658–1659. doi: 10.1093/bioinformatics/btl158
PubMed Abstract | Полный текст перекрестной ссылки | Академия Google
Лян, М., Рэйли, К., Чжэн, X., Катти, Г., Гогинени, Э., Шерман, Б. Т., и другие. (2016). Различение очень похожих изоформ генов с помощью кластерного биоинформатического анализа длинных чтений одиночных молекул PacBio. Биоданные мин. 9:13. doi: 10.1186/s13040-016-0090-8
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
Лиен С., Куп Б. Ф., Сандве С. Р., Миллер Дж. Р., Кент М. П., Ном Т. и др. (2016). Геном атлантического лосося дает представление о редиплоидизации. Природа 533, 200–205. doi: 10.1038/nature17164
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
Лю С., Чжан Ю., Чжоу З., Вальдбисер Г., Сунь Ф., Лу Дж. и др. (2012). Эффективная сборка и аннотация транскриптома сома с помощью анализа RNA-Seq удвоенной гаплоидной гомозиготы. BMC Genomics 13:595. doi: 10.1186/1471-2164-13-595
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
Лав, М. И., Хубер, В., и Андерс, С. (2014). Модерированная оценка изменения кратности и дисперсии для данных секвенирования РНК с помощью DESeq2. Геном Биол. 15:550. doi: 10.1186/s13059-014-0550-8
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
Маккуин, Д. Дж., и Джонстон, И. А. (2014). Хорошо ограниченная оценка времени дупликации всего генома лососевых выявляет значительное отделение от видовой диверсификации. Проц. биол. науч. 281:20132881. doi: 10.1098/rspb.2013.2881
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
Мартин, М. (2011). Cutadapt удаляет последовательности адаптеров из операций высокопроизводительного секвенирования. EMBnet J. 17:10. doi: 10.14806/ej.17.1.200
CrossRef Полный текст | Google Scholar
Маклафлин, М.Ф., и Грэм, Д.А. (2007). Альфавирусные инфекции у лососевых — обзор. Дж. Фиш Дис. 30, 511–531. doi: 10.1111/j.1365-2761.2007.00848.x
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
Наттестад М., Гудвин С., Нг К., Баслан Т., Седлазек Ф.Дж., Решенедер П. и др. (2018). Сложные перестройки и амплификации онкогенов, обнаруженные с помощью секвенирования длинной ДНК и РНК клеточной линии рака молочной железы. Рез. генома. 28, 1126–1135. doi: 10.1101/gr.231100.117
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
NCBI (2015). NCBI Salmo salar Annotation Release 100 Assembly Report [Online]. Доступно в Интернете по адресу: https://www.ncbi.nlm.nih.gov/genome/annotation_euk/Salmo_salar/100/ (по состоянию на 26 июня 2020 г.)
Google Scholar
Oikonomopoulos, S., Bayega, A., Fahiminiya , С., Джамбазян, Х., Берубе, П., и Рагуссис, Дж. (2020). Методологии профилирования транскриптов с использованием давно читаемых технологий. Фронт. Жене. 11:606. doi: 10.3389/fgene.2020.00606
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
PacBio (2020). IsoSeq v3: Масштабируемое открытие изоформ De Novo [онлайн]. Доступно в Интернете по адресу: https://github.com/PacificBiosciences/IsoSeq (по состоянию на 2020 г.). , SA (2020). Гибридный подход к секвенированию транскриптома улучшил сборку и аннотацию генов в Cynara cardunculus (L. ). BMC Genomics 21:317. doi: 10.1186/s12864-020-6670-5
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
Роадс А. и Ау К. Ф. (2015). Секвенирование PacBio и его приложения. Геном. протеом. биоинф. 13, 278–289. doi: 10.1016/j.gpb.2015.08.002
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
Робертс, Р. Дж., Карнейро, М. О., и Шац, М. К. (2013). Преимущества секвенирования SMRT. Геном Биол. 14:405. doi: 10.1186/gb-2013-14-6-405
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
Сахрейян С.М.Э., Мохиюддин М., Себра Р., Тилгнер Х., Афшар П.Т., Ау К.Ф. и др. (2017). Получение всестороннего биологического понимания транскриптома путем выполнения анализа секвенирования РНК широкого спектра. Нац. коммун. 8:59. doi: 10.1038/s41467-017-00050-4
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
Салмела Л. и Ривалс Э. (2014). LoRDEC: точное и эффективное исправление ошибок длительного чтения. Биоинформатика 30, 3506–3514. doi: 10.1093/bioinformatics/btu538
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
Шве А., Остбай Т.К., Краснов А., Рамберг С. и Андреассен Р. (2020). Характеристика дифференциально экспрессируемых миРНК и их предполагаемых транскриптов-мишеней во время смолтификации и адаптации к морской воде в головной почке атлантического лосося. Гены (Базель) 11:1059. doi: 10.3390/genes110
PubMed Abstract | Полный текст перекрестной ссылки | Академия Google
Смит А., Хабли Р. и Грин П. (2013). RepeatMasker Open-4.0. [Онлайн]. Доступно в Интернете по адресу: http://repeatmasker.org/ (по состоянию на 2018 г.). и другие. (2015). Смертность и потеря веса атлантического лосося Salmon salar L., экспериментально инфицированного изолятами лососевого альфавируса подтипа 2 и подтипа 3 из Норвегии. Дж. Фиш Дис. 38, 1047–1061. doi: 10.1111/jfd.12312
Резюме PubMed | Полный текст перекрестной ссылки | Google Scholar
Tardaguila, M., de la Fuente, L., Marti, C., Pereira, C., Pardo-Palacios, F.J., Del Risco, H., et al. (2018). SQANTI: обширная характеристика последовательностей длинных транскриптов для контроля качества при идентификации и количественном определении полноразмерного транскриптома. Рез. генома. 28, 396–411. doi: 10.1101/gr.222976.117
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
Тедерсоо Л., Туминг-Клундеруд А. и Анслан С. (2018). Метабаркодирование PacBio грибов и других эукариот: ошибки, предубеждения и перспективы. Новый Фитол. 217, 1370–1385. doi: 10.1111/nph.14776
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
Ценг, Э. (2020a). кДНК_Cupcake [онлайн]. Доступно в Интернете по адресу: https://github.com/Magdoll/cDNA_Cupcake (по состоянию на 2020 г.)
Google Scholar
Ценг, Э. (2020b). Cogent: Инструмент реконструкции генома COding [онлайн]. Доступно в Интернете по адресу: https://github.com/Magdoll/Cogent (по состоянию на 2020 г.)
Google Scholar
Ван Ю., Лю Х., Чжэн Д., Ван Ю., Чен Х., Чжао Х. и др. (2019). Систематическая идентификация межгенных длинных некодирующих РНК в сетчатке мыши с использованием полноразмерного секвенирования изоформ. BMC Genomics 20:559. doi: 10.1186/s12864-019-5903-y
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
Ван Б., Ценг Э., Регулски М., Кларк Т. А., Хон Т., Цзяо Ю. и др. (2016). Выявление сложности транскриптома кукурузы с помощью долговременного секвенирования одной молекулы. Нац. коммун. 7:11708. doi: 10.1038/ncomms11708
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
Волдемариам Н. Т., Агафонов О., Хойхайм Б., Хьюстон Р. Д., Таггарт Дж. Б. и Андреассен Р. (2019). Расширение репертуара микроРНК у атлантического лосося; открытие IsomiRs и микроРНК, высоко экспрессируемых в разных тканях и на разных стадиях развития. Ячейки 8:42. doi: 10.3390/cells8010042
PubMed Abstract | Полный текст перекрестной ссылки | Академия Google
Волдемариам, Н. Т., Агафонов, О., Синдре, Х., Хойхайм, Б., Хьюстон, Р. Д., Робледо, Д., и др. (2020). миРНК, которые, как предполагается, регулируют пути антивирусных генов хозяина у мальков атлантического лосося, зараженных IPNV, подвержены влиянию вирусной нагрузки и связаны с основными генотипами QTL устойчивости к IPN на поздних стадиях инфекции. Перед. Иммунол. 11:2113. doi: 10.3389/fimmu.2020.02113
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
Янез, Дж. М., Хьюстон, Р. Д., и Ньюман, С. (2014). Генетика и геномика устойчивости к болезням у видов лососевых. Фронт. Жене. 5:415. doi: 10.3389/fgene.2014.00415
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
Zhang, G., Sun, M., Wang, J., Lei, M., Li, C., Zhao, D., et al. (2019). Полноразмерное секвенирование кДНК PacBio, интегрированное с чтением RNA-seq, значительно улучшает обнаружение транскриптов сплайсинга в рисе. Завод J. 97, 296–305. doi: 10.1111/tpj.14120
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
Жулидов П. А. (2004). Простая нормализация кДНК с использованием дуплекс-специфичной нуклеазы камчатского краба. Рез. нуклеиновых кислот. 32:e37. doi: 10.1093/nar/gnh031
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
© 2004–2021 ISO, Unicode, Inc. |
Расшифровка транскриптов очень похожих мультигенных семейств из данных Iso-Seq с помощью IsoCon
Введение
Значительная часть генов в геноме человека принадлежит к мультигенным семействам, каждое из которых содержит несколько копий генов, возникших в результате дупликации, т.е. дубликатов генов 1,2,3,4,5,6 . Многие из этих дубликатов генов связаны с важными фенотипами человека, включая ряд заболеваний 7,8,9 . В некоторых из таких случаев отдельные копии генов играют различную роль в этиологии заболевания 10 . Однако аннотация мультигенных семейств остается неполной даже в самой последней сборке человека, особенно из-за неразрешенных сегментных дупликаций с высокой идентичностью последовательностей 11,12 . Дублированные копии генов из одного и того же семейства различаются по идентичности последовательностей, причем некоторые из них идентичны друг другу. Кроме того, количество копий внутри семей часто различается у разных людей 1,2,3 . Кроме того, по оценкам, >90% всех мультиэкзонных генов подвергаются альтернативному сплайсингу у людей 13,14 , а различные дублирующиеся копии генов могут различаться в продуцируемых альтернативно сплайсированных формах (т.е. изоформах).
Эти особенности делают расшифровку сквозных последовательностей транскриптов из дублированных генов и их различных изоформ транскриптов сложной задачей. Количество копий мультигенных семейств можно определить с помощью микрочипов 2 , количественная полимеразная цепная реакция (ПЦР) 15 , капельная цифровая ПЦР 16 или секвенирование ДНК с использованием Nanostring Technologies 8 или платформы Illumina 3 . Последовательности отдельных экзонов, длина которых составляет всего несколько сотен нуклеотидов, могут быть получены из отдельных прочтений данных Illumina DNA или RNA-seq 17 ; однако повторяющийся характер копий двойных генов усложняет их сборку de novo, а чтения Illumina часто неспособны фазировать варианты по длине полного транскрипта и имеют низкую скорость отзыва в сборках генов с множественными изоформами 18,19 . Длинные чтения Pacific Biosciences (PacBio) из протокола Iso-Seq обладают потенциалом для преодоления этой проблемы путем последовательного секвенирования множества транскриптов. Этот подход был успешно применен для выявления нескольких сложных структур изоформ, возникающих в результате событий альтернативного сплайсинга, например, у людей, растений и грибов 18,20,21 . Ни в одном из этих исследований одновременно не рассматривались проблемы расшифровки структуры изоформ и определения копий генов, из которых они произошли.
Несмотря на то, что частота ошибок PacBio снизилась, многие ошибки по-прежнему трудно исправить, и они остаются серьезной проблемой для последующего анализа данных Iso-Seq 19,22,23 . Это особенно касается транскриптов из семейств генов с высокой идентичностью последовательностей, где трудно выделить ошибки из истинных вариантов. Использование эталонного генома 24,25,26,27,28 для коррекции неэффективно в таких ситуациях, когда изменчивость копий гена не может быть надежно зафиксирована эталоном. ЛЕД 18 , часть биоинформатического конвейера PacBio для обработки данных Iso-Seq, является стандартным инструментом, используемым для исправления ошибок секвенирования без использования эталона. Хотя ICE использовался в нескольких проектах 29,30 , было показано, что он генерирует большое количество избыточных расшифровок 18,21,31 . Более того, ICE «в настоящее время не настроен для работы по дифференциации очень сложных семейств генов от полиплоидных видов, где различия в основном основаны на SNP». 32 . Альтернативный подход — использовать считывания Illumina для исправления ошибок в считываниях PacBio 25,26,28,33 — также неспособен исправить большинство ошибок (как мы демонстрируем в этой статье) и также имеет смещение из-за низкой глубины считывания Illumina в GC. богатые регионы 34 .
Были предложены некоторые подходы для исправления ошибок чтения PacBio из транскриптов с высокой идентичностью последовательностей, но ни один из них не применим в широком смысле для определения последовательностей из мультигенных семейств с высокой идентичностью последовательностей без опоры на эталонный геном. Классификация 35 или конструирование 36 аллель-специфических транскриптов с Iso-Seq были описаны, но эти подходы требуют ссылки и могут разделять только два аллеля одного гена. Подходы к генотипированию для мультигенных семейств также были предложены 37,38 , но они требуют предварительного знания последовательностей изоформ. Подход de novo для кластеризации очень похожих изоформ описан в ref. 39 , но реализация не предусмотрена. Проблема также связана с вирусной фазой 9.0058 40 , но методы, разработанные там, не применимы напрямую к мультигенным семьям.
Еще одним соображением является относительно высокая стоимость PacBio. Количество прочтений, необходимых для восстановления семейств генов, экспрессия которых затмевается суперраспространенными классами мРНК, может быть непомерно высокой. Подход к целевому секвенированию может быть эффективным для сокращения необходимого объема секвенирования, когда пары праймеров ОТ-ПЦР предназначены для извлечения транскриптов интересующего семейства генов 41 . Этот подход приводит к достаточно высокой глубине секвенирования, чтобы захватить большинство расшифровок и выполнить последующую коррекцию ошибок.
Чтобы устранить эти ограничения, мы разрабатываем IsoCon, алгоритм de novo для исправления ошибок и устранения избыточности считываний циклической согласованной последовательности (CCS) PacBio, созданных в результате целевого секвенирования с помощью протокола Iso-Seq. Наш алгоритм позволяет расшифровывать последовательности изоформ вплоть до уровня нуклеотидов и выдвигать гипотезы о том, как они относятся к отдельным, очень похожим копиям генов мультигенных семейств. IsoCon использует осторожный итеративный процесс для исправления очевидных ошибок без чрезмерного исправления редких вариантов. Его статистическая структура предназначена для использования возможностей длинных чтений для связывания вариантов в расшифровке. Кроме того, IsoCon статистически учитывает большую изменчивость качества чтения, которая имеет тенденцию к снижению по мере увеличения длины стенограммы. Используя смоделированные данные, мы демонстрируем, что IsoCon имеет значительно более высокую точность и полноту, чем ICE 9.0058 18 в широком диапазоне глубины секвенирования, а также длин транскриптов, сходств и уровней распространенности.
Мы применяем IsoCon для изучения семейств ампликонных генов Y-хромосомы, где невозможность изучения отдельных копий генов и их соответствующих транскриптов ограничила наше понимание эволюции Y-хромосомы приматов и причин нарушений мужского бесплодия, для которых эти гены имеют решающее значение 42,43,44,45 . Семейства ампликонных генов Y-хромосомы представляют собой особенно интересный и сложный случай для расшифровки, поскольку каждое из них содержит несколько почти идентичных (до 99,99%) копий 46,47 с потенциально различным числом изоформ. Мы используем целенаправленный дизайн для выделения и секвенирования транскриптов всех девяти семейств ампликонических генов Y-хромосомы из яичек двух мужчин. Наша проверка показывает, что IsoCon значительно повышает точность по сравнению с исправлением ошибок на основе ICE и Illumina с proovread 48 и имеет значительно более высокую полноту, чем ICE. Мы показываем, что IsoCon может обнаруживать редкие транскрипты, которые отличаются всего на одну пару оснований от доминирующих изоформ, которые имеют на два порядка большее количество. Используя предсказанные IsoCon транскрипты, мы можем зафиксировать беспрецедентное количество изоформ, отсутствующих в существующих базах данных. Кроме того, мы можем разделить транскрипты на предполагаемые копии генов и получить специфичные для копии последовательности экзонов и варианты сплайсинга.
Чтобы продемонстрировать более широкую применимость IsoCon, мы также запустили его на общедоступном наборе данных целевого секвенирования Iso-Seq гена FMR1 49 . FMR1 является членом семейства генов, связанных с ломкой Х-хромосомой, и отвечает за развитие как синдрома ломкой Х-хромосомы, так и синдрома тремора/атаксии, ассоциированного с ломкой Х-хромосомой, нейродегенеративного расстройства, возникающего у взрослых. FMR1 подвергается обширному альтернативному сплайсингу, который был предметом нескольких исследований 49 . Используя IsoCon, мы можем восстановить больше изоформ, чем ICE, и найти новые кандидаты на сплайс-соединения. Наши результаты показывают, что разница в количестве изоформ между носителями и контролем не так велика, как сообщалось ранее в ref. 49 .
Результаты
Моделированные данные
Мы создали синтетические семейства генов, используя три эталонных гена в качестве исходной последовательности для нашего моделирования: TSPY , HSFY и ДАЗ . Мы выбрали их, потому что они отражают спектр длины, числа экзонов и сложности, характерный для семейств ампликоновых генов Y (таблица 1). DAZ представляет собой наиболее сложный случай, так как он имеет сильно повторяющуюся экзонную структуру 50 . Длина гена также важна, поскольку более длинные транскрипты приводят к меньшему количеству проходов полимеразы во время секвенирования и, следовательно, к более высокой частоте ошибок чтения CCS. Мы смоделировали уровни покрытия в диапазоне, согласующемся с тем, что мы наблюдали в реальных данных.
Полноразмерная таблица
Наше основное моделирование было сосредоточено на двух сценариях. Первый (рис. 1) отражает типичный биологический сценарий. Для каждого из трех семейств генов мы моделировали несколько копий гена и для каждой копии моделировали различные изоформы, пропуская разные экзоны. В каждом семействе было всего 30 смоделированных изоформ, абсолютная распространенность которых была определена случайным образом из значений 2 i , i ∈[1,8], что приводит к относительной численности от 0,1% до 15%. Мы генерируем три таких набора данных, варьируя частоту мутаций, используемую для создания дублирующихся копий генов. (Обратите внимание, что здесь для простоты мы моделируем только мутацию, хотя известно, что другие процессы, например конверсия генов, влияют на эволюцию дубликатов копий генов 51 .) Второе моделирование (дополнительный рисунок 1) похоже на первое, но , чтобы отделить эффект мутации от эффекта пропуска экзона, мы не моделируем изоформы. Для каждого семейства генов было смоделировано в общей сложности восемь копий генов и восемь транскриптов (по одному на копию гена) с различной идентичностью последовательностей (дополнительный рисунок 2) и с относительным содержанием в диапазоне от 0,4% до 50%. Мы также повторили эти два моделирования, но сохранили постоянное содержание изоформ (дополнительные рисунки 3 и 4). Полное описание нашей симуляции см. в дополнительном примечании 2.
Полнота и точность для транскриптов с разной экзонной структурой и неодинаковой долей. Скрипичные графики, показывающие отзыв ( a ) и точность ( b ) IsoCon и ICE. На каждой панели строки соответствуют разным семействам и, следовательно, разным частотам ошибок. Самое короткое семейство генов ( TSPY , помеченное по имени копии референсного гена, использованной для его создания, TSPY13P ) с соответственно более низким уровнем ошибок чтения показано в верхних строках панели, в то время как самое длинное семейство генов ( DAZ , обозначенный как DAZ2 ), с соответственно более высоким уровнем ошибок чтения, показан в нижних строках. Столбцы соответствуют разным частотам мутаций ( μ ), используемым при моделировании копий генов (см. Дополнительное примечание 2). Более низкая частота мутаций означает большее количество одинаковых копий генов. На каждом графике показаны результаты для 30 изоформ со случайным распределением содержания в диапазоне от 0,1% до 15%. На каждом графике ось x соответствует количеству смоделированных считываний, а y — ось показывает полноту/точность методов. Каждая скрипка создается с использованием 10 смоделированных повторов секвенирования. Белая точка показывает медиану, толстая черная линия — межквартильный размах (средние 50%), более тонкая черная линия — 95% доверительный интервал, а цветная область — график плотности. Мы отмечаем, что график плотности обрезается в самых крайних точках данных
Полноразмерное изображение
Точность IsoCon увеличивается с увеличением глубины чтения, даже когда средняя глубина чтения на расшифровку достигает 1562x (дополнительный рисунок 1). Такой надежности часто трудно достичь, потому что увеличение охвата сверх того, что необходимо для отзыва, только увеличит количество ошибок в данных. Отзыв зависит от частоты ошибок, на которую влияет длина гена. Для TSPY , при равных показателях численности отзыв становится идеальным при 17-кратном охвате, в то время как для DAZ отзыв достигает> 90% только при 410-кратном охвате (дополнительный рисунок 3). Мы ожидаем, что точность также будет зависеть от сходства копий гена, т. е. семейство генов, которое создается с использованием низкой скорости мутаций, тем самым производя меньше вариантов между копиями гена, может негативно повлиять на способность IsoCon разделять транскрипты. Несколько удивительно, но точность в этих случаях снижается лишь незначительно, а глубина считывания оказывает гораздо более существенное влияние на точность, чем частота мутаций или длина гена.
Наши эксперименты ясно показывают, что припоминание IsoCon сильно зависит от глубины чтения. Мы исследовали это более подробно, взяв каждый транскрипт, смоделированный как часть экспериментов на дополнительном рисунке 1. Мы показываем (рис. 2), захватывал ли IsoCon транскрипт в зависимости от глубины секвенирования (т. Е. Общее количество прочтений) его соответствующий эксперимент и его собственная глубина секвенирования (т.е. количество прочтений, которые были секвенированы из изоформы). Для TSPY , IsoCon фиксирует большинство транскриптов с глубиной >3, в то время как это число составляет ~10 для HSFY . Скорость мутации играет лишь незначительную роль по сравнению с глубиной транскрипта, поскольку большинство кандидатов с меньшей глубиной чтения теряются на этапе исправления ошибок (рис. 2). Мы также наблюдаем, что для DAZ минимальная глубина транскрипта, необходимая для захвата изоформы, увеличивается по мере увеличения общей глубины секвенирования, предполагая, что относительное количество транскриптов также является фактором. Вероятно, это связано с тем, что матрица множественного выравнивания становится все более шумной (особенно для DAZ ), поскольку количество последовательностей растет, что негативно влияет как на исправление ошибок, так и на расчет поддержки в статистическом тесте.
Возможности IsoCon для записи расшифровок. Каждая изоформа из экспериментов на дополнительном рисунке 1 (включая 10 смоделированных повторов) соответствует маркеру на этом графике, который отмечен в зависимости от того, был ли он захвачен и выведен как окончательный прогноз (зеленый), полученный на этапе исправления ошибок, но отфильтрованный. отсутствует на статистическом шаге (синий) или вообще не производится на этапе коррекции (красный) с помощью IsoCon. Это полоса, созданная с помощью пакета Seaborn 9.0058 64 , который представляет собой особый тип точечной диаграммы, где ось x является категориальной (общее количество прочтений соответствующего эксперимента), а точки распределены по горизонтали. Ось y показывает количество прочтений, секвенированных из изоформы, в логарифмической шкале. Изоформы, у которых нет прочтений, не показаны
Полноразмерное изображение
Мы также наблюдаем, что IsoCon превосходит ICE как по точности, так и по полноте (рис. 1, дополнительные рисунки 1, 3–5). ICE имеет низкую точность, которая снижается с увеличением глубины чтения. Например, при глубине чтения 500 и выше точность ICE близка к 0 во всех наших экспериментах. С другой стороны, точность IsoCon всегда >80% при глубине считывания 500 и выше. IsoCon также имеет более высокий отзыв почти во всех случаях для HSFY и DAZ . Что касается TSPY , преимущество отзыва колеблется между двумя алгоритмами, но в целом довольно похоже. Дальнейшее исследование производительности IsoCon подробно описано в дополнительном примечании 3.
Данные из двух образцов семенников человека
Мы получили данные целевого секвенирования транскриптов RT-PCR для девяти семейств ампликоновых генов для двух образцов семенников мужчин, используя протокол Iso-Seq от PacBio. (Дополнительный рисунок 6 показывает количество проходов за чтение) и отдельно с использованием технологии секвенирования Illumina. Затем мы использовали конвейер ToFU 9.0058 18 , чтобы отфильтровать любые считывания PacBio CCS, которые либо были химерными, либо не охватывали весь транскрипт от конца до конца. Полученный набор мы называем исходным (CCS) чтением. Для сравнения мы запустили IsoCon и ICE 18 при чтении CCS. Мы также оценили инструмент proovread 48 , который использует чтения Illumina для исправления показаний CCS (называемых Illumina-скорректированными чтениями CCS). Дополнительное примечание 4 содержит подробные сведения о том, как эти инструменты запускались. Мы сравнили результаты трех подходов, а также подхода с использованием только исходных считываний CCS. В таблице 2 показано количество сгенерированных прочтений и количество транскриптов, вызванных каждым из этих четырех различных подходов.
Полноразмерная таблица
Валидация
Для проверки IsoCon и сравнения его точности с другими методами мы использовали (1) показания Illumina, (2) внутреннюю согласованность между образцами и (3) соответствие с базой данных эталонных транскриптов.
Мы подтвердили точность на уровне нуклеотидов показаний IsoCon, ICE, CCS с поправкой на Illumina и исходных показаний с помощью данных Illumina, полученных для тех же двух человек. Во всех положениях в предсказанных транскриптах мы классифицировали положение как поддерживаемое, если оно имело по крайней мере два прочтения Illumina, совпадающих с ним по тому же нуклеотиду, что и транскрипт. Поскольку глубина секвенирования Illumina была на несколько порядков выше, чем у PacBio (таблица 2), мы ожидаем, что будет поддерживаться большинство правильных позиций. Обратите внимание, что отсутствие поддержки Illumina не всегда указывает на ошибку, поскольку смещение Illumina GC приведет к тому, что некоторые регионы не секвенируются. Однако мы ожидаем, что количество ошибок расшифровки будет коррелировать с количеством неподдерживаемых позиций. На рис. 3 показаны проценты поддерживаемых нуклеотидов для каждого подхода. В среднем 9Поддерживается 9% позиций транскриптов IsoCon, но поддерживаются только 93% позиций транскриптов ICE. Точно так же поддерживаются 96% скорректированных Illumina позиций чтения и 79% исходных позиций чтения. Кроме того, 70% расшифровок IsoCon полностью поддерживаются (т. е. в каждой отдельной позиции) Illumina, по сравнению с 2% для ICE, 15% для прочтений, скорректированных Illumina, и 20% для нескорректированных прочтений.
Illumina-поддержка предсказанных транскриптов. Гистограмма показывает процентное соотношение позиций в IsoCon/ICE/корректном чтении и исходных считываниях CCS flnc, которые поддерживаются как минимум двумя выравниваниями Illumina с одним и тем же нуклеотидом
Полноразмерное изображение
Хотя мы ожидаем некоторую изменчивость в транскриптах, присутствующих в двух образцах, мы также ожидаем, что большая их часть будет общей. IsoCon обнаружил 121 транскрипт, который присутствует в обоих образцах, что соответствует 32% общего количества транскриптов (в среднем между двумя образцами; дополнительная фигура 7). Чтения CCS, скорректированные Illumina, разделяют 11%, в то время как и ICE, и исходные чтения разделяют менее 2%. Вероятно, это указывает на более высокую точность IsoCon по сравнению с другими методами.
IsoCon также лучше справился с восстановлением известных расшифровок Ensembl. Мы загрузили аннотированные кодирующие последовательности девяти семейств ампликонических генов Y-хромосомы из базы данных Ensembl 52 , содержащую 61 уникальный транскрипт после удаления избыточности (см. Дополнительное примечание 5). Затем мы идентифицировали стенограммы базы данных, которые полностью соответствовали предсказанным стенограммам. У IsoCon было 21 совпадение с Ensembl, в то время как у ICE было только восемь (включая совпадения IsoCon; рис. 4). IsoCon также имел больше совпадений с базой данных, чем исходные чтения, несмотря на уменьшение количества последовательностей в> 29 раз.(Таблица 2). CCS-прочтения с поправкой Illumina в целом имели на одно точное совпадение больше, чем IsoCon, но имели более чем в 14 раз больше предсказанных транскриптов IsoCon, что свидетельствует о низкой точности.
Способность методов фиксировать эталонные кодирующие белок транскрипты в базе данных ENSEMBL. Цифры в скобках рядом с названием семейства генов на оси x указывают количество уникальных транскриптов в базе данных. Кодирующие последовательности отсутствуют для XKRY
Полноразмерное изображение
Мы также исследовали точность транскриптов IsoCon, которые имели более высокие значения p значимости, и обнаружили, что, хотя точность немного снизилась по сравнению с транскриптами с более низкими значениями p , она все же остается значительно выше, чем Чтения CCS с поправкой на ICE или Illumina (дополнительное примечание 5).
Разнообразие изоформ
Для изучения транскриптов, обнаруженных IsoCon, мы сначала отфильтровали транскрипты, обнаруженные только в одном образце. Хотя мы ожидаем изменчивости между двумя людьми, такие транскрипты также могут быть ложноположительными, возникающими из-за ошибок ПЦР с обратной транскриптазой (ОТ-ПЦР) 53 . Эти ошибки, если они присутствуют, были введены до создания библиотеки и могут быть обнаружены как при чтении Iso-Seq, так и при чтении Illumina. Они приведут к уникальным последовательностям, которые будут имитировать настоящие транскрипты как в данных Iso-Seq, так и в данных Illumina. В нашем последующем анализе используется только 121 транскрипт, который был одинаково предсказан IsoCon в обоих образцах. Это устраняет любые ошибки ОТ-ПЦР, присутствующие только в одном образце, и уменьшает количество ложноположительных транскриптов из-за неисправленных ошибок секвенирования. В таблице 3 показано количество общих транскриптов, разделенных на семейства генов. Однако мы отмечаем, что истинное количество транскриптов в образце может быть выше из-за специфичных для образца вариантов, которые мы отбросили. Далее мы классифицировали каждый транскрипт как белок-кодирующий или некодирующий в зависимости от того, находится ли он в кадре или вне кадра с эталонными транскриптами человека (подробности см. в Дополнительном примечании 5). Мы обнаружили, что 72 из 121 транскрипта являются кодирующими, а пять из девяти семей содержат в общей сложности 49 транскриптов.некодирующие транскрипты (таблица 3), остальные четыре семейства имеют только кодирующие транскрипты. Мы также обнаружили, что 94 из 121 транскрипта не были известны ранее (таблица 3), т. е. не имели 100% совпадения, охватывающего весь транскрипт, при сопоставлении с базой данных неизбыточных нуклеотидов NCBI ( nr / nt ). Множественное выравнивание для транскриптов IsoCon RBMY — семейства с наиболее предсказанными транскриптами — показано на рис. 5.
Новые транскрипты — это те, которые не имеют идеального соответствия справочной базе данных транскриптов NCBI, числа в скобках указывают дополнительные транскрипты, которые имеют идеальное соответствие только с EST, синтетическими конструкциями или транскриптами, предсказанными in silico Полноразмерная таблица
Рис. 5 Иллюстрация взаимосвязи между 61 транскриптом RBMY, предсказанным IsoCon и общим для обоих образцов. Транскрипты пронумерованы от 1 до 61. В левой части рисунка используется IGV 65,66 для визуализации множественного выравнивания транскриптов. Закрашенные позиции — это позиции с изменчивостью в транскриптах, тогда как серые области обозначают консервативные позиции. Удаления показаны горизонтальной линией с номером, указывающим их длину. Правая часть рисунка иллюстрирует взаимосвязь между 61 стенограммой в виде графика. Вершины являются транскриптами (помечены их индексами). Вершина выделена жирным шрифтом, если предполагается, что она кодирует белок. Ребро между двумя транскриптами означает, что они являются потенциальными изоформами из одной и той же копии гена, т. е. у них есть различия только в наличии/отсутствии экзонов. Чтобы упростить визуализацию, некоторые вершины окружены прямоугольниками, а двойное ребро между двумя прямоугольниками указывает на то, что все пары транскриптов между двумя прямоугольниками являются потенциальными изоформами из одной и той же копии гена. Каждая максимальная клика (т. е. группа вершин), состоящая из более чем четырех вершин, показана в виде цветного круга. Цвета кружков соответствуют строкам множественного выравнивания, отмеченным вертикальной чертой того же цвета. Максимальную клику следует интерпретировать как все транскрипты, которые потенциально происходят из одной и той же копии гена 9.0003
Полноразмерное изображение
IsoCon чувствителен к небольшим вариациям и вариациям с низким содержанием
IsoCon смог обнаружить несколько транскриптов даже в присутствии изоформы с гораздо более высоким содержанием, которые отличались всего на 1–3 п. н. Например, один транскрипт RBMY , извлеченный IsoCon в образце 2, поддерживался только пятью прочтениями и отличался только одним нуклеотидом от транскрипта, который поддерживался 863 прочтениями. Второй пример — еще один IsoCon RBMY 9.0497, который поддерживался только пятью прочтениями в образце 2 и отличался только одним нуклеотидом от транскрипта, который поддерживался 306 прочтениями. Оба этих транскрипта с более низким содержанием были получены в обоих образцах (поддержка этих транскриптов в образце 1 составляла 10 и 9 прочтений соответственно), имели идеальную поддержку Illumina и кодировали белок. Ни один из них не был обнаружен ICE или представлен без ошибок в исходных чтениях; однако оба они также были получены в считываниях CCS, скорректированных Illumina. На рис. 5 показаны эти два транскрипта с меньшей распространенностью, обозначенные цифрами 3 и 1 соответственно.
Разделение транскриптов на копии генов
Семейство генов состоит из копий генов, каждая из которых может образовывать несколько изоформ в результате альтернативного сплайсинга. В таких случаях транскрипты будут выравниваться друг с другом с большими вставками/делециями (из-за отсутствия экзонов), но без замен. Мы определяем минимальное количество групп (т. е. кластеров), необходимых для того, чтобы каждый транскрипт можно было отнести по крайней мере к одной группе, а каждая пара транскриптов в одной и той же группе отличалась только большими вставками/делециями (подробности см. в дополнительном примечании 5). . Мы называем это количеством групп, которое является нашей наилучшей оценкой количества копий, то есть размера каждого семейства генов. Обратите внимание, что аллель-специфические транскрипты не существуют для семейств ампликонных генов Y из-за гаплоидной природы Y-хромосомы. Мы также определяем количество групп кодирования, то есть количество групп, рассчитанное только из транскриптов кодирования. Группа соответствует понятию максимальной клики из теории графов 54 , а поскольку количество расшифровок относительно невелико, количество групп можно вычислить с помощью алгоритма грубой силы (дополнительное примечание 5).
Количество групп для девяти различных семейств показано в таблице 3, а на рис. 5 показано представление о группах на примере семейства RBMY . Важное различие между группой и копией гена заключается в том, что транскрипт может принадлежать нескольким группам. Это происходит, если экзон, содержащий вариант, разделяющий две копии гена, пропускается в процессе сплайсинга. В таком случае мы не можем определить, из какой копии происходит стенограмма, и наш подход помещает ее в обе группы. Таким образом, размер каждой группы является оценкой сверху количества изоформ, происходящих из каждой копии гена.
Мы отмечаем, что истинное количество копий в семействе генов может быть выше или ниже количества групп, определенных IsoCon, по нескольким причинам. Во-первых, невозможно разделить транскрипты, происходящие от копий с идентичными экзонными последовательностями. В результате мы можем недооценить истинное количество копий. Во-вторых, число копий может отличаться у двух проанализированных самцов 55 . Поскольку мы исключаем транскрипты, уникальные для каждого самца, мы можем недооценить истинное количество копий. Кроме того, количество копий для семейства генов может быть одинаковым у двух мужчин, но некоторые из копий могут иметь разные последовательности. В-третьих, могут быть копии, которые биологически различаются только наличием/отсутствием экзонов или другими большими вставками — если нет замен, полученные транскрипты будут сгруппированы вместе и будут рассматриваться как происходящие из одной и той же копии гена в соответствии с нашим подходом. Поскольку большинство ампликонических генов Y-хромосомы человека были образованы путем дупликации целой области 56 , такая ситуация не должна быть распространена. Тем не менее, если бы он присутствовал, это занижало бы количество копий. В-четвертых, редактирование РНК может генерировать транскрипты, имеющие замены, но происходящие из одной и той же копии, что приводит к переоценке истинного числа копий. В-пятых, наш подход к групповым транскриптам основан на точности выравнивания транскриптов, которые иногда могут быть неточными при наличии повторов. В-шестых, наш подход иногда помещает транскрипт более чем в одну группу (как описано выше) и, следовательно, может завышать количество изоформ, происходящих из каждой копии гена. Учитывая эти предостережения, мы, тем не менее, ожидаем, что количество групп будет полезным показателем размера генного семейства.
Мы сравнили наше количество кодирующих групп с количеством копий, аннотированных на Y-хромосоме в эталонном геноме человека (GRCh48/hg38), и наблюдалось в предыдущих исследованиях вариаций ДНК в человеческих популяциях (таблица 3). Для одного из семейств генов ( HSFY ) мы не обнаружили общих транскриптов между людьми. Для четырех семейств генов ( CDY , DAZ , RBMY и TSPY ) количество кодирующих групп находится в пределах ранее наблюдаемого диапазона на основе анализа ДНК в популяциях человека (таблица 3). Для оставшихся четырех семейств генов ( BPY , PRY , VCY и XKRY ), количество кодирующих групп меньше, чем количество копий, указанное в предыдущих исследованиях. Три из этих семей — BPY, VCY и XKRY— имели только один кодирующий транскрипт, общий для двух образцов. Таким образом, в целом количество кодирующих групп является консервативной оценкой количества копий генов на семейство ампликоновых генов.
Новые варианты сплайсинга
Для 121 предсказания IsoCon, которые были разделены между образцами, 38 транскриптов указывали на новые вариации в координатах начала интрон-экзон (дополнительная таблица 1, подробности анализа в дополнительном примечании 4). Это включает 21 расшифровку «новый в каталоге» (определено в ссылке 9).0058 57 как «содержащие новые комбинации ранее известных сплайс-соединений или новых сплайс-соединений, образованных из уже аннотированных доноров и акцепторов»), и 17 транскриптов, содержащих по крайней мере одно сплайс-соединение, которое не совпадало с каким-либо известным сплайс-соединением (соответствующим либо новое соединение сплайсинга или к копии гена, не представленной на hg19). Все 21 новые в каталоге транскрипты имели сильную поддержку Illumina (по крайней мере, четыре высококачественных полноразмерных выравнивания Illumina) через соединения сплайсов и выровнены по hg19.без замен или вставок вблизи стыков (что дает нам уверенность в выравнивании вблизи стыков). Из остальных 17 транскриптов 15 имели сильную поддержку Illumina в отношении потенциально новых соединений сплайсинга.
Некоторые транскрипты отличались друг от друга только небольшими внутриэкзонными мутациями, но не паттерном сплайсинга. Таким образом, 38 транскриптов соответствуют 25 уникальным новым паттернам сплайсинга, из которых 13 представляют новые паттерны сплайсинга в каталоге. Из 25 10 кодируют, а 15 не кодируют (таблица 3).
Данные из
FMR1 гена Целевая Iso-Seq была выполнена в исх. 49 гена FMR1 у трех носителей (премутационные носители синдрома тремора/атаксии, ассоциированного с ломкой Х-хромосомой), каждый из которых секвенирован с тремя клетками SMRT, и трех контролей, каждый из которых секвенирован с одной клеткой SMRT. Конвейер ToFU (использующий ICE для кластеризации и исправления ошибок) использовался в сочетании с выравниванием эталонного генома для получения всего 49 изоформ, которые присутствовали по крайней мере в одном из шести образцов. Три дополнительные изоформы были предсказаны в более раннем исследовании 58 , но в исх. 49 . Мы используем эти 52 изоформы для оценки производительности IsoCon. Обратите внимание, что Ценг и его коллеги в первую очередь интересовались структурой сплайсинга и не изучали точечные мутации или вставки. Поэтому мы не анализировали эти аспекты результатов IsoCon. Подробности анализа см. в дополнительном примечании 6.
При рассмотрении присутствия/отсутствия 49 изоформ в каждом из образцов IsoCon обнаружил в среднем 24 изоформы на образец (дополнительная таблица 3), в то время как Ценг и его коллеги обнаружили только 20 (Дополнительная таблица 9в 49 ). IsoCon также обнаружил две из трех предсказанных изоформ из 58 , которые не были обнаружены в исх. 49 . Из 24 изоформ, существование которых было предсказано Претто и его коллегами для этого гена, общее число подтвержденных составляет 23.
Одно из интересных открытий исх. 49 заключается в том, что из 46 изоформ, обнаруженных у носителей, 30 не были обнаружены ни в одном из контрольных образцов. Ценг и его коллеги предположили потенциальную роль этого в вредной активности белка. Однако IsoCon смог обнаружить 5 из этих 30 в контроле. Хотя большинство изоформ по-прежнему были специфичны для носителей, наши результаты показывают, что разница может быть меньше, чем обнаруженная в исх. 49 .
Многие изоформы, обнаруженные IsoCon, но не Ценгом и его коллегами, присутствовали при низком охвате (дополнительная таблица 3). Например, пять дополнительных транскриптов, обнаруженных IsoCon в контроле, имели небольшое количество (3–9) прочтений, поддерживающих их. В целом мы заметили, что IsoCon имел лучшую относительную эффективность в обнаружении изоформ для контрольных образцов, которые были секвенированы на одной трети глубины случаев. В частности, для трех контролей IsoCon обнаружил в среднем 17 изоформ на образец, в то время как Ценг и его коллеги обнаружили только 12 (по сравнению с носителями, где среднее количество обнаруженных изоформ составляло 31 и 27 соответственно). Наши результаты показывают, что IsoCon более чувствителен при меньшем покрытии, чем ICE.
Кроме того, разрешение IsoCon на уровне нуклеотидов полезно для обнаружения новых сайтов сплайсинга. Например, мы обнаружили новый вариант сплайсинга, принадлежащий к изоформам группы C (как классифицировано в ссылке 49 ). IsoCon обнаружил вариант сплайсинга у всех трех носителей премутации и у одного из контролей. Транскрипт, содержащий новое соединение сплайсинга, отличается от ранее полученной изоформы 7 в группе C наличием сайта сплайсинга, начинающегося пятью парами оснований ниже по течению до начала экзона 17, см. Дополнительную фигуру 8a. В то время как Набор данных FMR1 не имеет прочтений Illumina для подтверждения соединения сплайсинга, есть несколько факторов, позволяющих предположить, что это достоверная изоформа 23 : новый сайт сплайсинга является каноническим (т. или замены в выравнивании с hg19, и он имеет высокую поддержку чтения CCS (> 105 в каждом из трех носителей премутации). Точно так же мы наблюдали новое соединение сплайсинга в результате делеции последнего нуклеотида в экзоне 11, которое присутствовало в четырех изоформах в группах C и D и поддерживалось тысячами прочтений CCS (дополнительный рисунок 8b). Такие прогнозы, созданные IsoCon, могут стать кандидатами для дальнейших исследований.
Обсуждение
Мы использовали как смоделированные, так и экспериментальные данные, чтобы продемонстрировать эффективность IsoCon при получении транскриптов из мультигенных семей с высокой идентичностью. Алгоритмически эту проблему можно рассматривать как обобщение получения аллель-специфических вариантов гена. Однако в IsoCon не реализована функциональность, которая позволяла бы отличать аллель-специфические варианты от отдельных копий гена. Однако мы считаем, что IsoCon подходит для получения аллель-специфических транскриптов. В таком сценарии потребуется дальнейший дальнейший анализ, если кто-то захочет отличить разные аллели от разных копий.
Используемые здесь экспериментальные методы имеют некоторые потенциальные ограничения. Во-первых, Iso-Seq может не захватить очень мало транскриптов, поскольку глубокое секвенирование PacBio может быть непомерно дорогим. Это ограничение потенциально может быть преодолено путем дополнения данных Iso-Seq данными Illumina RNA-seq и модификации IsoCon для включения таких данных. Во-вторых, наш подход, состоящий только в секвенировании транскриптома, не дает окончательного ответа на вопрос о размере каждого семейства генов и не позволяет нам окончательно отнести каждый транскрипт к копии гена. Точно так же трудно отличить новые копии генов от вариантов сплайсинга или редактирования РНК. Чтобы облегчить это, можно амплифицировать и секвенировать экзоны одного и того же человека и сравнивать последовательности, полученные из РНК и ДНК. В-третьих, хотя мы видим очень хорошую поддержку Illumina расшифровок IsoCon, мы не можем исключить возможность других источников ошибок, таких как ошибки RT-PCR. Чтобы решить эту проблему, из одного и того же образца могут быть приготовлены две библиотеки репликации кДНК, хотя потребуются дальнейшие проверки. Наконец, подход обогащения мишеней на основе ПЦР, который мы использовали здесь, мог не захватить транскрипты копий генов с мутациями в сайтах праймеров ПЦР. Альтернативой этому подходу является использование метода захвата на основе зонда 9.0058 59 , который не зависит от праймеров для ПЦР.
Несмотря на эти ограничения, IsoCon позволил нам обнаружить беспрецедентное количество изоформ, многие из которых являются новыми, а также получить более точные оценки количества копий генов в семействах ампликоновых генов Y. IsoCon также может быть полезен для расшифровки изоформ генов со значительным альтернативным сплайсингом, таких как FMR1 . IsoCon также чувствителен к незначительным смещениям в местах сращивания. Например, три RBMY -транскрипты (19, 31 и 33 на рис. 5) представляли собой сплайс-варианты, отличающиеся только 3–5-нуклеотидной разницей в сплайсинге с ближайшим совпадающим транскриптом; IsoCon обнаружил аналогичные варианты в данных FMR1 (дополнительный рисунок 8). Хотя такие прогнозы все еще нуждаются в подтверждении, они позволяют проводить дальнейший функциональный анализ и, как ожидается, откроют новые возможности для исследования Y-ампликоновых генов.
Методы
Обзор IsoCon
Входными данными для алгоритма IsoCon являются наборы ридов PacBio CCS по крайней мере с одним полным проходом транскрипта и их базовые прогнозы качества. IsoCon предполагает, что чтения были предварительно обработаны с помощью биоинформатического конвейера Iso-Seq для удаления праймеров, штрих-кодов и прочтений, которые являются химерами или не охватывают весь транскрипт. На этапе предварительной обработки чтения разделяются в соответствии с парами праймеров, используемых для амплификации отдельных семейств генов, и IsoCon запускается отдельно для каждого семейства генов. Результатом IsoCon является набор расшифровок, которые являются результатом исправления ошибок чтения и отчета о каждом отдельном чтении.
IsoCon состоит из двух основных этапов: (i) алгоритм итеративной кластеризации для исправления ошибок считываний и идентификации транскриптов-кандидатов и (ii) итеративное удаление статистически незначимых кандидатов.
Этап кластеризации/исправления разделяет операции чтения на кластеры, где операции чтения, похожие друг на друга, объединяются в один кластер. Множественное выравнивание и консенсусная последовательность вычисляются для каждого кластера. Чтения в каждом кластере затем частично исправляются до согласованной последовательности кластера; чтобы избежать удаления истинных вариантов, исправляется только половина потенциально ошибочных столбцов. Затем процесс повторяется — измененные операции чтения перераспределяются в потенциально разные кластеры и снова корректируются. Этот процесс повторяется до тех пор, пока в каком-либо кластере не перестанут обнаруживаться различия, а оставшиеся отдельные последовательности называются транскриптами-кандидатами (или просто кандидатами).
Этап кластеризации/исправления спроектирован так, чтобы быть чувствительным, и поэтому за ним следует второй этап, на котором удаляются транскрипты-кандидаты, недостаточно поддерживаемые исходными (неисправленными) считываниями. Первоначально исходные чтения назначаются одному из их ближайших совпадающих кандидатов. Затем, оценивая все пары близких кандидатов, для каждой пары мы проверяем, есть ли достаточные доказательства того, что назначенные им прочтения на самом деле не происходят из одного и того же транскрипта. Для этого мы берем двух кандидатов и набор вариантов позиций (т. е. позиций, в которых два кандидата различаются) и формулируем тест гипотезы, чтобы сделать вывод, насколько вероятно, что чтения, поддерживающие эти варианты, вызваны ошибками секвенирования. Поскольку кандидат может участвовать во многих попарных тестах, ему присваивается наименее значимый p — значение из всех выполненных парных тестов. После проверки всех пар кандидатов часть незначимых кандидатов будет удалена. Затем повторяется второй этап IsoCon — исходные чтения назначаются наилучшему совпадению из оставшихся кандидатов, которые затем подвергаются статистической проверке. Это продолжается до тех пор, пока все оставшиеся кандидаты не станут значимыми. Затем оставшиеся кандидаты выводятся как предсказанные стенограммы.
Кластеризация и исправление ошибок, шаг
Во-первых, нам нужно определить концепцию ближайших соседей и графа ближайших соседей. Пусть dist( x , y ) обозначает расстояние редактирования между двумя строками x и y . Пусть S будет мультимножеством строк. Для строки x мы говорим, что y ∈ S является ближайшим соседом x в S , если \({\mathrm {dist}}\left( {x,y} \right ) = \ mathop {{{\ mathrm {min}}}} \ limit_ {z \ in S} {\ mathrm {dist}} \ left ({x, z} \ right) \). то есть y имеет наименьшее расстояние до x в S . Граф ближайших соседей S — это ориентированный граф, вершинами которого являются строки S , и существует ребро от x до y тогда и только тогда, когда y является ближайшим соседом х , но не х .
Этап кластеризации/исправления состоит из двух фаз — фазы разделения и фазы коррекции — и мы итерируемся между фазами. На этапе разбиения мы сначала разбиваем чтения на кластеры, при этом каждый кластер имеет ровно одно чтение, обозначаемое как центр. Идея состоит в том, что каждый раздел содержит предполагаемый набор прочтений, происходящих из одной и той же расшифровки, а центром является прочтение, последовательность которого наиболее похожа на последовательность расшифровки. Для разбиения сначала построим граф G , который изначально идентичен графу ближайших соседей, построенному на основе чтений. Затем мы идентифицируем чтение x в G с наибольшим количеством вершин, которое может достигать x . Мы создаем новый кластер с x в качестве центра и содержащий все чтения в G , которые имеют путь к x , включая сам x . Затем мы удаляем элементы нового кластера вместе с инцидентными им ребрами из G . Затем мы повторяем только что модифицированные G : определение вершины с наибольшим количеством вершин, которые могут ее достичь, и создание кластера вокруг нее. Полный псевдокод приведен в алгоритме PartitionStrings на дополнительном рис. 9.
Результирующий раздел обладает тем свойством, что каждая строка имеет одного из ближайших соседей (не включая себя) в своем кластере. Этот ближайший сосед может быть центром, но не обязательно. Таким образом, кластер может содержать множество строк, являющихся ближайшими соседями других, но только одна из них обозначается как центральная.
Фаза коррекции работает независимо с каждым кластером ридов и соответствующим ему центром. Сначала мы создаем попарные выравнивания от каждого чтения к центру, используя parasail 60 . Затем мы создаем матрицу множественного выравнивания A из попарных выравниваний (подробности см. в дополнительном примечании 1). Каждая запись в A является либо нуклеотидом, либо пробелом, и каждая строка соответствует чтению. Мы получаем консенсус A , беря наиболее часто встречающийся символ в каждом столбце. Каждая клетка в 9t\), а поддержка ячейки в A — это поддержка состояния этой ячейки в столбце этой ячейки. Поддержка зависит от состояния, чтобы быть более чувствительным к различным типам ошибок. Например, поскольку часто встречаются делеции и вставки, для того, чтобы эти варианты не исправлялись, требуется больший охват по сравнению с заменой. Затем при каждом чтении мы идентифицируем вариантные позиции (т. е. состоянием которых является замена, вставка или удаление) и выбираем половину этих позиций с наименьшей поддержкой. Затем для каждой из этих позиций исправляем ее на самый частый символ в столбце; но если наиболее часто встречающийся символ не уникален, то исправление не производится.
Этап кластеризации/коррекции IsoCon сочетает этапы разделения и коррекции следующим образом. Сначала мы разбиваем набор чтений и исправляем каждый кластер. Говорят, что кластер сошелся, если все его строки идентичны. Пока хотя бы один кластер не сошелся, мы повторяем этапы разбиения и исправления. Чтобы гарантировать, что в конце концов все кластеры сойдутся, мы эвристически отменяем исправление строки, если после исправления она имеет большее расстояние редактирования до центра, чем было до своего центра в предыдущей итерации, если строка чередуется между разбиениями в циклическом цикле. образом, или один и тот же набор строк повторно назначается одному и тому же разделу, где они различаются только в позициях, где наиболее часто встречающийся символ не определен. Наконец, после того, как все разделы сойдутся, мы назначаем их центры кандидатами в транскрипты и переходим к этапу фильтрации кандидатов IsoCon. Полный псевдокод для этого шага приведен в подпрограмме ClusterCorrect на дополнительном рисунке 9..
Шаг фильтрации кандидатов
Второй шаг IsoCon принимает в качестве входных данных набор ридов X и набор расшифровок-кандидатов \(C = \{ c_1, \ldots ,c_l\}\). Первым шагом является назначение операций чтения кандидатам таким образом, чтобы одна операция чтения была назначена ровно одному из ближайших соседей-кандидатов в C . Поскольку чтение может иметь несколько ближайших соседей-кандидатов в C , существует много возможных назначений. Для наших целей мы используем следующий итерационный жадный алгоритм. За каждое чтение x ∈ X , мы идентифицируем его ближайших соседей-кандидатов в C . Затем мы выбираем кандидата c ∈ C , который является ближайшим соседом большинства чтений в X . Мы присваиваем все эти чтения c и удаляем c из C и все назначенные чтения из X . Затем мы повторяем процесс, используя уменьшенные X и C , пока не будут назначены все чтения.
Теперь у нас есть назначение чтений кандидатам. Обозначим через X i чтения, назначенные кандидату c i . Мы проверяем доказательства, подтверждающие, что c i является верным кандидатом, следующим образом. Мы рассматриваем кандидатов, которые являются ближайшими соседями c i в \(C — \{ c_i\}\). Затем для каждого ближайшего соседа-кандидата c j мы формируем нулевую гипотезу о том, что считывания в X i and in X j originated from c j , i. e. c i is not a true candidate. Расчет значения значимости при этой нулевой гипотезе приведен в следующем разделе. Мы вычисляем p i , наименьшее значимое значение среди всех c j . Мы ограничиваем наши сравнения c i только своим ближайшим соседям-кандидатам, потому что это сохраняет эффективность нашего алгоритма и маловероятно, что сравнение с другими, более непохожими кандидатами увеличит p i .
Затем мы идентифицируем кандидатов с p i выше порога значимости α . Это α является параметром нашего алгоритма, установленным по умолчанию на 0,01. Затем эти кандидаты удаляются из набора кандидатов 9.0496 С . Учитывая параметр τ , если имеется более τ кандидатов со значением значимости выше α , мы удаляем только τ лучших кандидатов с самыми высокими значениями. Затем выполняется этап фильтрации кандидатов алгоритма: мы снова назначаем чтения кандидатам и идентифицируем кандидатов с недостаточной поддержкой в соответствии с нашей проверкой гипотезы. Алгоритм останавливается, когда больше нет кандидатов с p i выше α . Псевдокод для этого алгоритма вместе со всем IsoCon приведен на дополнительном рисунке 9. и X d , которые были присвоены им. Мы используем \(x_i \in X_c \cup X_d\) для обозначения каждого чтения, и пусть n будет количеством чтений в \(X_c \cup X_d\). Мы вычисляем попарные выравнивания от \(X_c \cup X_d \cup \{c\}\) до д . Затем мы строим матрицу множественного выравнивания A из этих попарных выравниваний так же, как и на этапе коррекции (подробности см. в Дополнительном примечании 1). Каждая запись в A соответствует либо нуклеотиду, либо пробелу. Пусть V будет индексом столбцов A , где c и d не совпадают. Мы называем эти позиции вариантными позициями.
Лет А i,j обозначают символ в столбце j строки i строки A . Строки \(1 \le i \le n\) соответствуют чтениям x i , а строка n + 1 соответствует c . Для \(1 \le i \le n\) мы определяем двоичную переменную S i , которая равна 1 тогда и только тогда, когда n +1, j для всех j ∈ V . То есть, S I IS 1, если и только тогда, когда прочитал x I , поддерживает все варианты V , I.E. E. Share As C V , I.E. E. Share As C V , т.е. А . Мы делаем следующие предположения:
- 1.
d , действующая в качестве эталонной последовательности в этом тесте, не содержит ошибок.
- 2.
Нуклеотид в прочтении в позиции, которой нет в V и отличается от соответствующего нуклеотида в d , вызван ошибкой секвенирования. Другими словами, в положении, когда c и d совпадают, они не могут быть оба неправильными.
- 3.
Вероятности ошибки в двух разных позициях чтения независимы.
- 4.
S i и \(S_{i\prime}\) являются независимыми случайными величинами для всех \(i \ne i\prime\).
Наша нулевая гипотеза состоит в том, что вариантные позиции в A возникают из-за ошибок последовательности в X . Чтобы получить распределение S i в соответствии с нулевой гипотезой, нам сначала нужна вероятность, обозначаемая p ij , что позиция j при чтении i соответствует i . ошибка. В значительной степени это можно получить из оценок качества Phred в чтении (подробности см. в Дополнительном примечании 1). При предположении 3 имеем, что S i следует распределению Бернулли со средним значением \(p_i = \mathop {\prod}\limits_{j \in V} {p_{ij}}\).
Релевантная тестовая статистика при нулевой гипотезе — это количество, которое моделирует силу (или значимость) поддержки вариантов V . Мы хотели бы подсчитывать только чтения, которые полностью поддерживают все варианты, т. е. чтения x i с s i = 1( s
7 7 9533 i обозначает наблюдаемое значение S i ). Эти чтения могут иметь ошибки в невариантных местоположениях, но в вариантных местоположениях они должны согласовываться с c . Для каждого такого прочтения мы хотели бы взвесить его вклад обратной величиной вероятности того, что все символы в различных местоположениях вызваны ошибками последовательности. Интуитивно, чтение с высоким базовым качеством должно считаться большим доказательством, чем чтение с низким базовым качеством. Принимая во внимание эти соображения, мы определяем нашу тестовую статистику как 9{с_{я}}}}\)
Обратите внимание, что p i уменьшается с количеством вариантов в V и с более высокими базовыми показателями качества; следовательно, \(T\) предназначен для использования связанных вариантов в расшифровке, в том смысле, что требуется меньше чтений для поддержки расшифровки, когда расшифровка имеет больше вариантов. Более того, \(p_i\) уменьшается для прочтений с более высоким базовым качеством CCS в вариантных позициях, что означает, что для поддержки транскрипта требуется меньше прочтений, если они имеют более высокое качество. Мы заметили, что значения качества оснований в CCS сильно варьируют и зависят от (i) количества проходов в прочтении CCS, (ii) длины мононуклеотида и (iii) секвенированного основания, с C и G имеющие более низкие качества, связанные с ними (дополнительный рис. 10)
Пусть \(t\) будет наблюдаемым значением этой статистики, и мы будем называть его взвешенной поддержкой. Учитывая \(t\), мы вычисляем значение значимости как \(P(T \ge t)\). Мы используем односторонний тест, поскольку нас интересуют только значения значимости равной или более взвешенной поддержки для \(V\). Нам неизвестно о закрытой форме распределения \(T\) при нулевой гипотезе, и грубый подход к вычислению \(P(T \ge t)\) был бы невозможен. Однако мы можем воспользоваться следующей теоремой из ref. 9п {\ гидроразрыва {{p_i \ log \, p_i}} {{\ mathop {{{\ mathrm {max}}}} \ limits_k \ left ({ — \ log \, p_k} \ right)}}} \)
Обратите внимание, что при этом преобразовании \(P\left( {T\prime \ge t\prime } \right) = P\left( {T \ge t} \right)\), поскольку логарифмическая функция строго монотонно, а нормализация с использованием максимума постоянна. \mu \)
Мы используем эту верхнюю границу в качестве значения значимости. Обратите внимание, что теорема применима только для \(\delta > 0\). Если \(t\prime \le \mu \), то это не так. Однако это означает, что наблюдаемая взвешенная поддержка ниже ожидаемой поддержки в соответствии с нулевой гипотезой. Такие значения явно незначительны, и наше программное обеспечение по умолчанию использует значение 0,5. Транскрипты-кандидаты, которые имеют более порогового значения вариантов позиций (по умолчанию 10) по сравнению со всеми другими транскриптами-кандидатами, статистически не оцениваются, поскольку их p -значение будет близко к 0.
Отношение к ICE
IsoCon, как и ICE, использует итеративный кластерный и консенсусный подход, но эти два алгоритма имеют фундаментальные различия. После кластеризации IsoCon получает взвешенный консенсус на основе профиля ошибок в разделе и использует его в качестве информации для исправления ошибок чтения; ICE, с другой стороны, получает консенсус кластера, используя автономный вызывающий консенсус DAGCON 62 , который будет использоваться в следующей итерации без исправления ошибок чтения. IsoCon и ICE также различаются графами, которые они используют для моделирования отношений между последовательностями, и алгоритмом разделения графа на кластеры. IsoCon детерминистически создает кластеры, моделируемые как задача обхода пути, в то время как ICE моделирует кластер как максимальную клику и использует недетерминированный аппроксимативный алгоритм максимальной клики. Возможно, наиболее важно то, что IsoCon, в отличие от ICE, включает в себя статистическую структуру, которая позволяет отличать ошибки от истинных вариантов с более высокой точностью.
Экспериментальные методы
Поли(А) РНК была выделена из РНК семенников двух мужчин европеоидной расы (идентификаторы: CR560016, возраст 59 лет, образец 1; CR561118, возраст 79 лет, образец 2; Origene) с использованием набора Poly(A) Purist MAG ( Термо Фишер Сайентифик). 50 нг поли(А) РНК на каждый образец вместе с 1 мкг контрольной тотальной РНК печени (использовали для контроля) использовали для создания двухцепочечной ДНК с использованием набора для синтеза кДНК SMARTer PCR (Clontech). Была проведена оптимизация цикла ПЦР реакции амплификации кДНК с использованием праймера Clontech, и было определено, что 12 циклов являются оптимальными для крупномасштабной ПЦР-амплификации. Для каждого из девяти семейств ампликоновых генов мы разработали пару праймеров для ОТ-ПЦР, причем один праймер расположен в первом, а другой — в последнем кодирующем экзоне (дополнительная таблица 4). Для одного из этих семейств генов ( CDY ), была разработана дополнительная пара праймеров для захвата транскриптов, происходящих из всех копий гена (дополнительная рис. 11). Один из двух уникальных штрих-кодов PacBio был добавлен к праймерам, чтобы различать продукты ОТ-ПЦР у двух мужчин. Затем продукты ОТ-ПЦР от этих двух человек были разделены на два эквимолярных пула в соответствии с ожидаемыми размерами транскриптов (<1 kb и 1–2 kb; дополнительная таблица 4) и очищены с использованием гранул AMPure XP (Beckman Coulter, Inc., США). ). Затем каждый из двух пулов ОТ-ПЦР использовали для создания отдельной библиотеки PacBio Iso-Seq, которая была секвенирована с помощью RSII (химия P6-C4) с использованием одной клетки SMRT на библиотеку. Таким образом, всего было секвенировано две клетки SMRT.
Кроме того, мы секвенировали те же продукты ОТ-ПЦР с помощью технологии Illumina. Мы создали отдельную библиотеку Nextera XT (с уникальной парой индексов) для каждой комбинации пара праймеров и образцов. Всего было проанализировано девять семейств генов с использованием 10 пар праймеров (как упоминалось выше, одно семейство генов, CDY , было проанализировано с двумя парами праймеров). Таким образом, было сконструировано 10 пар праймеров × 2 человека = 20 библиотек. Эти библиотеки были нормализованы, объединены в эквимолярном соотношении и секвенированы на приборе MiSeq с использованием одного набора MiSeq Reagent Nano Kit, v2 (секвенирование парных концов 250 × 250).
Расширенная версия экспериментального протокола доступна в Интернете по адресу https://doi.org/10.1038/protex.2018.109.
Доступность кода
IsoCon имеет открытый исходный код и находится в свободном доступе по адресу https://github. com/ksahlin/IsoCon. Результаты IsoCon в этой статье были получены с коммитом 79589f3 на GitHub. Подробная информация о параметрах программного обеспечения приведена в дополнительном примечании 4. Скрипты для всех анализов доступны по адресу https://github.com/ksahlin/IsoCon_Eval. Этот репозиторий также включает в себя змейку 63 рабочий процесс, который воспроизводит промежуточные и окончательные данные для анализа семейства ампликоновых генов.
Ссылки
Заррей, М., Макдональд, Дж. Р., Мерико, Д. и Шерер, С. В. Карта вариаций числа копий генома человека. Нац. Преподобный Жене. 16 , 172–183 (2015).
КАС Статья Google ученый
Картер, Н. П. Методы и стратегии анализа вариаций числа копий с использованием ДНК-микрочипов. Нац. Жене. 39 , С16–С21 (2007).
КАС Статья Google ученый
Редон, Р. и др. Глобальная вариация числа копий в геноме человека. Природа 444 , 444–454 (2006).
ОБЪЯВЛЕНИЯ КАС Статья Google ученый
Бейли, Дж. А., Кидд, Дж. М. и Эйхлер, Э. Э. Полиморфные гены числа копий человека. Цитогенет. Геном Res. 123 , 234–243 (2008).
КАС Статья Google ученый
Ли, У.-Х., Гу, З., Ван, Х. и Некрутенко, А. Эволюционный анализ генома человека. Природа 409 , 847–849 (2001).
ОБЪЯВЛЕНИЯ КАС Статья Google ученый
Брахмачари, М. и др. Цифровое генотипирование макросателлитов и мультикопийных генов выявляет новые биологические функции, связанные с изменением числа копий больших тандемных повторов. PLoS Genet. 10 , e1004418 (2014).
Артикул Google ученый
Конрад Б. и Антонаракис С. Э. Дублирование генов: стремление к фенотипическому разнообразию и причина болезней человека. год. Преподобный Геном. Гум. Жене. 8 , 17–35 (2007).
КАС Статья Google ученый
Цучия Н., Кёгоку К., Мияшита Р. и Куроки К. Разнообразие мультигенных семейств иммунной системы человека и его влияние на генетический фон ревматических заболеваний.
Курс. Мед. хим. 14 , 431–439 (2007).КАС Статья Google ученый
Бейли, Дж. А., Явор, А. М., Масса, Х. Ф., Траск, Б. Дж. и Эйхлер, Э. Э. Сегментные дупликации: организация и влияние в текущей сборке проекта генома человека. Рез. генома. 11 , 1005–1017 (2001).
КАС Статья Google ученый
Бейли, Дж. А. и Эйхлер, Э. Э. Сегментарные дупликации приматов: тигли эволюции, разнообразие и болезни. Нац. Преподобный Жене. 7 , 552–564 (2006).
КАС Статья Google ученый
Пан, К., Шай, О., Ли, Л.Дж., Фрей, Б.Дж. и Бленкоу, Б.Дж. Глубокое исследование сложности альтернативного сплайсинга в транскриптоме человека с помощью высокопроизводительного секвенирования. Нац. Жене. 40 , 1413–1415 (2008).
КАС Статья Google ученый
Wang, E. T. et al. Альтернативная регуляция изоформ в транскриптомах тканей человека. Природа 456 , 470–476 (2008).
ОБЪЯВЛЕНИЯ КАС Статья Google ученый
Хигути Р., Доллинджер Г., Шон Уолш П. и Гриффит Р. Одновременная амплификация и обнаружение специфических последовательностей ДНК. Биотехнология 10 , 413–417 (1992).
КАС Статья Google ученый
Hindson, B.J. et al. Высокопроизводительная цифровая система капельной ПЦР для абсолютного количественного определения количества копий ДНК. Анал. хим. 83 , 8604–8610 (2011).
КАС Статья Google ученый
Steijger, T.
et al. Оценка методов реконструкции транскриптов для секвенирования РНК. Нац. Методы 10 , 1177–1184 (2013).КАС Статья Google ученый
Гордон С.П. и др. Широко распространенные полицистронные транскрипты у грибов, обнаруженные с помощью секвенирования одиночных молекул мРНК. PLoS ONE 10 , e0132628 (2015).
Артикул Google ученый
Лю, X., Мей, В., Солтис, П. С., Солтис, Д. Э. и Барбазук, В. Б. Обнаружение альтернативно сплайсированных изоформ транскриптов из одномолекулярных долгочитаемых последовательностей без эталонного генома. Мол. Экол. Ресурс. 17 , 1243–1256 (2017).
КАС Статья Google ученый
Чжан, С.-Дж. и другие. Эволюция изоформ у приматов посредством независимой комбинации альтернативных событий процессинга РНК.
Мол. биол. Эвол. 34 , 2453–2468 (2017).КАС Статья Google ученый
Hoang, N.V. et al. Обзор сложного транскриптома из высокополиплоидного генома сахарного тростника с использованием полноразмерного секвенирования изоформ и сборки de novo из короткого секвенирования. BMC Геном. 18 , 395 (2017).
Артикул Google ученый
Kuo, R. I. et al. Нормализованное секвенирование длинной РНК у курицы выявило сложность транскриптома, подобную человеческой. BMC Геном. 18 , 323 (2017).
Артикул Google ученый
Tardaguila, M. et al. SQANTI: обширная характеристика последовательностей длинного считывания транскриптов для контроля качества при идентификации и количественном определении полноразмерного транскриптома.
https://doi.org/10.1101/118083 (2017 г.).Abdel-Ghany, S.E. et al. Обзор транскриптома сорго с использованием длинных чтений одиночных молекул. Нац. коммун. 7 , 11706 (2016).
ОБЪЯВЛЕНИЯ КАС Статья Google ученый
Weirather, J.L. et al. Характеристика генов слияния и значительно выраженных изоформ слияния при раке молочной железы с помощью гибридного секвенирования. Рез. нуклеиновых кислот. 43 , e116 (2015).
Артикул Google ученый
Донг, Л. и др. Секвенирование транскриптов отдельных молекул в реальном времени облегчает аннотацию генома мягкой пшеницы и исследование транскриптома зерна. BMC Геном. 16 , 1039 (2015).
Артикул Google ученый
Minoche, A. E. et al. Использование секвенирования одномолекулярных транскриптов для предсказания эукариотических генов. Геном Биол. 16 , 184 (2015).
Артикул Google ученый
Ченг, Б., Фуртадо, А. и Генри, Р. Дж. Длинное чтение транскриптома кофейных зерен показывает разнообразие полноразмерных транскриптов. Gigascience 6 , 1–13 (2017).
Артикул Google ученый
Ван, Б. и др. Выявление сложности транскриптома кукурузы с помощью долговременного секвенирования одной молекулы. Нац. коммун. 7 , 11708 (2016).
ОБЪЯВЛЕНИЯ КАС Статья Google ученый
Workman, R. E. et al. Полноразмерное секвенирование одной молекулы дает представление об экстремальном метаболизме колибри с красным горлом Archilochus colubris. https://doi.org/10.1101/117218 (2017 г.).
PacificBiosciences. PacificBiosciences/кДНК_праймер. Гитхаб . https://github.com/PacificBiosciences/cDNA_primer/wiki/RS_IsoSeq-(v2.3)-Tutorial-%232.-Isoform-level-clustering-(ICE-and-Quiver). По состоянию на 16 ноября 2017 г.
Au, K.F. et al. Характеристика транскриптома ЭСК человека с помощью гибридного секвенирования. Проц. Натл акад. науч. США 110 , E4821–E4830 (2013 г.).
КАС Статья Google ученый
Лав, М. И., Хогенеш, Дж. Б. и Иризарри, Р. А. Моделирование смещения последовательности фрагментов РНК-секвенции снижает систематические ошибки в оценке распространенности транскриптов.
Нац. Биотехнолог. 34 , 1287–1291 (2016).КАС Статья Google ученый
Тилгнер, Х., Груберт, Ф., Шэрон, Д. и Снайдер, М.П. Определение персонального, аллель-специфического и одномолекулярного долгочитаемого транскриптома. Проц. Натл акад. науч. США 111 , 9869–9874 (2014).
ОБЪЯВЛЕНИЯ КАС Статья Google ученый
Мангул, С. и др. HapIso: точный метод реконструкции изоформ, специфичных для гаплотипов, из длинных одномолекулярных ридов. https://doi.org/10.1101/050906 (2016).
Нуманагич, И. и др. Cypiripi: точное генотипирование CYP2D6 с использованием данных высокопроизводительного секвенирования. Биоинформатика 31 , i27–i34 (2015).
Артикул Google ученый
Лян, М. и др. Различение очень похожих изоформ генов с помощью кластерного биоинформатического анализа длинных чтений одиночных молекул PacBio. Мин. биоданных 9 , 13 (2016).
Артикул Google ученый
Артёменко А. и др. Длинные чтения одной молекулы могут разрешить сложность вируса гриппа, состоящего из редких, близкородственных мутантных вариантов. Дж. Вычисл. биол. 24 , 558–570 (2017).
КАС Статья Google ученый
Чжан, В., Циклитира, П. и Мессинг, Дж. PacBio секвенирование семейств генов — тематическое исследование с генами глютена пшеницы.
Ген 533 , 541–546 (2014).КАС Статья Google ученый
Сков, Л., Датский пангеномный консорциум и Шируп, М. Х. Анализ 62 собранных гибридов Y-хромосом человека выявил быстрые структурные изменения и высокую скорость конверсии генов. Генетика PLoS. 13 , e1006834 (2017).
Артикул Google ученый
Ахмади Растегар, Д. и др. Профили экспрессии генов на уровне изоформы генов фактора азооспермии Y-хромосомы человека и их паралогов X-хромосомы в ткани яичка пациентов с необструктивной азооспермией. J. Proteome Res. 14 , 3595–3605 (2015).
КАС Статья Google ученый
Giachini, C. et al. TSPY 1 Изменение числа копий влияет на сперматогенез и демонстрирует различия между линиями Y.
Дж. Клин. Эндокринол. Метаб. 94 , 4016–4022 (2009).КАС Статья Google ученый
Ферлин, А., Моро, Э., Гаролла, А. и Фореста, С. Мужское бесплодие человека и делеции Y-хромосомы: роль генов-кандидатов AZF DAZ , RBM и DFFRY. Гул. Воспр. 14 , 1710–1716 (1999).
КАС Статья Google ученый
Бхоумик Б.К., Сатта Ю. и Такахата Н. Происхождение и эволюция семейств ампликоновых генов человека и ампликоновой структуры. Рез. генома. 17 , 441–450 (2007).
КАС Статья Google ученый
Скалецкий Х. и др. Специфичная для мужчин область Y-хромосомы человека представляет собой мозаику дискретных классов последовательностей. Природа 423 , 825–837 (2003).
ОБЪЯВЛЕНИЕ КАС Статья Google ученый
Tseng, E., Tang, H.-T., AlOlaby, R.R., Hickey, L. & Tassone, F. Измененная экспрессия ландшафта вариантов сплайсинга FMR1 у носителей премутаций. Биохим. Биофиз. Acta 1860 , 1117–1126 (2017).
КАС Статья Google ученый
Громолл, Дж. и др. Ген обезьяны Старого Света DAZ (удаленный в AZoospermia) дает представление об эволюции кластера генов DAZ на Y-хромосоме человека. Гул. Мол. Жене. 8 , 2017–2024 (1999).
КАС Статья Google ученый
Акен Б.Л. и др. Ensembl 2017. Nucleic Acids Res. 45 , Д635–Д642 (2017).
КАС Статья Google ученый
Фунгтаммасан, А. и др. Ошибки обратной транскрипции и различия РНК-ДНК при коротких тандемных повторах. Мол. биол. Эвол. 33 , 2744–2758 (2016).
КАС Статья Google ученый
Дистель, Р. Теория графов 5-е изд. (Спрингер, Берлин, 2018 г.).
Томашкевич, М. и др. Эффективная с точки зрения времени и затрат стратегия секвенирования Y-хромосом млекопитающих: приложение к сборке de novo Y-хромосомы гориллы.
Рез. генома. 26 , 530–540 (2016).КАС Статья Google ученый
Hurles, M. Дублирование генов: геномная торговля запчастями. PLoS Биол. 2 , E206 (2004 г.).
Артикул Google ученый
Tardaguila, M. et al. SQANTI: обширная характеристика последовательностей длинного считывания транскриптов для контроля качества при идентификации и количественном определении полноразмерного транскриптома. https://doi.org/10.1101/118083 (2017 г.).
Pretto, D. I. et al. Дифференциальное увеличение специфических изоформ мРНК FMR1 у носителей премутаций. J. Med. Жене. 52 , 42–52 (2015).
КАС Статья Google ученый
Альберт, Т.Дж. и др. Прямая селекция геномных локусов человека с помощью микрочиповой гибридизации.
Нац. Методы 4 , 903–905 (2007).КАС Статья Google ученый
Daily, J. Parasail: библиотека SIMD C для глобального, полуглобального и локального парного выравнивания последовательностей. БМС Биоинформ. 17 , 81 (2016).
Артикул Google ученый
Рагхаван П. Вероятностное построение детерминированных алгоритмов: аппроксимация целочисленных программ упаковки. Дж. Вычисл. Сист. науч. 37 , 130–143 (1988).
MathSciNet Статья Google ученый
Чин, К.-С. и другие. Негибридные, готовые сборки микробного генома из давно прочитанных данных секвенирования SMRT. Нац. Методы 10 , 563–569 (2013).
КАС Статья Google ученый
Майкл Васком и др. (2014, 14 ноября). seaborn: v0.5.0 (ноябрь 2014 г.) (версия v0.5.0). Зенодо. https://doi.org/10.5281/zenodo.12710. По состоянию на 22 ноября 2017 г.
Торвальдсдоттир, Х., Робинсон, Дж. Т. и Месиров, Дж. П. Integrative Genomics Viewer (IGV): высокопроизводительная визуализация и исследование данных геномики. Краткая информация. Биоинформ. 14 , 178–192 (2012).
Артикул Google ученый
Robinson, J. T. et al. Интегративный просмотрщик геномики. Нац. Биотехнолог. 29 , 24–26 (2011).
КАС Статья Google ученый
Сков, Л. Датский пангеномный консорциум и Шируп, М. Х. Анализ 62 собранных гибридов Y-хромосом человека выявил быстрые структурные изменения и высокую скорость генной конверсии. Генетика PLoS. 13 , e1006834 (2017).
Артикул Google ученый
Томашкевич, М. и др. Эффективная с точки зрения времени и затрат стратегия секвенирования Y-хромосом млекопитающих: приложение к сборке de novo Y-хромосомы гориллы. Рез. генома. 26 , 530–540 (2016).
КАС Статья Google ученый
Sudmant, P.H. et al. Разнообразие вариаций числа копий человека и мультикопийность генов. Наука 330 , 641–646 (2010).
ОБЪЯВЛЕНИЯ КАС Статья Google ученый
Ruderfer, D.M. et al. Паттерны генной непереносимости редких вариаций количества копий в 59 898 экзомах человека. Нац. Жене. 48 , 1107–1111 (2016).
КАС Статья Google ученый
Гао, С. и др. Профилирование полноразмерного транскриптома PacBio экспрессии митохондриальных генов насекомых. РНК Биол. 13 , 820–825 (2016).
Артикул Google ученый
Нуманагич, И. и др. Аллельная декомпозиция и точное генотипирование высокополиморфных и структурно-вариантных генов. Нац. коммун. 9 , 828 (2018).
ОБЪЯВЛЕНИЯ Статья Google ученый
Hackl, T., Hedrich, R., Schultz, J. & Förster, F. proovread: крупномасштабная высокоточная коррекция PacBio посредством итеративного консенсуса по краткому чтению. Биоинформатика 30 , 3004–3011 (2014).
КАС Статья Google ученый
Розен, С. и др. Обильная конверсия генов между плечами палиндромов в Y-хромосомах человека и обезьяны. Природа 423 , 873–876 (2003).
ОБЪЯВЛЕНИЯ КАС Статья Google ученый
Кестер, Дж. и Рахманн, С. Snakemake — масштабируемый механизм рабочего процесса в области биоинформатики. Биоинформатика 28 , 2520–2522 (2012).
Артикул Google ученый
Скалецкий Х. и др. Специфичная для мужчин область Y-хромосомы человека представляет собой мозаику дискретных классов последовательностей. Природа 423 , 825–837 (2003).
ОБЪЯВЛЕНИЯ КАС Статья Google ученый
Ссылки на скачивание
Стенограмма веб-семинара, который я провел по ISO 27035 Управление инцидентами безопасности
Недавно я провел учебный веб-семинар по реагированию на инциденты безопасности ISO 27035. Это стенограмма урока, который я вел. Пожалуйста, простите любые ошибки транскрипции. Майкл С. Редмонд
Примечания к стенограмме
- Доктор Майкл С. Редмонд, доктор философии MBCP, FBCI, CEM, PMP, MBA
- Киберзащита и реагирование Политика безопасности и средства контроля организации должны быть адаптированы к новым угрозам в современном мире. Оценка угроз безопасности продолжается и должна быть сопоставлена с адекватностью и наличием мер безопасности. Применяемые в настоящее время меры безопасности и контрмеры могут не соответствовать потенциальным рискам. Усилия никогда не заканчиваются, но главное знать, как начать.
- Мотиваторы Увеличение количества сообщаемых инцидентов компьютерной безопасности Увеличение числа и типов организаций, затронутых инцидентами компьютерной безопасности Более целенаправленное осознание организациями необходимости политик и методов безопасности в рамках их общих стратегий управления рисками Новые законы и правила, влияющие на то, как организации должны защищать информационные активы Осознание того, что системные и сетевые администраторы в одиночку не могут защитить организационные системы и активы
- Почему нарушения безопасности CRIST и последующее мошенничество становятся все более частыми и масштабными. В то время как финансовые учреждения, розничные торговцы, поставщики медицинских услуг и другие целевые организации делают все возможное, чтобы оставаться на шаг впереди киберпреступников, эти инциденты, вероятно, будут продолжаться, подвергая риску конфиденциальную информацию. Хотя вы не всегда можете предотвратить нарушение, быстрое реагирование может минимизировать ущерб для репутации и финансовые последствия. Проактивное и своевременное информирование владельцев счетов может помочь сократить расходы, в том числе связанные с увеличением активности колл-центра, обучением клиентов, кампаниями по восстановлению бренда, соблюдением нормативных требований и расходами на покрытие убытков клиентов.
- Многие крупные компании подвергаются хакерским атакам: Anthem, Sony, Target и другие. Количество утечек данных увеличилось на 27,5% в 2014 г. В компаниях все чаще принимаются меры против таких инцидентов безопасности.
- В 2012 г. в результате крупнейшей атаки на государственное агентство было украдено 3,8 миллиона налоговых документов • В августе хакеры украли номера социального страхования и кредитных карт из Департамента доходов Южной Каролины. Фишинговое электронное письмо позволило хакерам украсть учетные данные пользователей и в конечном итоге украсть 74 ГБ зашифрованных и незашифрованных данных.
- Взлом сервера в 2012 году привел к нарушению HIPAA Министерством здравоохранения штата Юта • В апреле 2012 года 780 000 человек пострадали от взлома сервера на уровне аутентификации, который позволил хакерам получить доступ и украсть SSN и личные медицинские записи из Министерства здравоохранения штата Юта. • Один сервер не был настроен в соответствии с обычной процедурой, что позволило хакерам получить доступ к системе.
- В 2012 году утечка данных PCI Global Payments Inc. затронула 1,5 миллиона человек • Почти 1,5 миллиона потребителей пострадали от хакеров, получивших доступ к системе обработки платежей Global Payments Inc. в январе и феврале.
- 14 декабря 2014 г. сбой в работе веб-сайта правительства Нидерландов из-за кибератаки • Кибер-злоумышленники вывели из строя основные веб-сайты правительства Нидерландов в течение большей части вторника, а резервные планы оказались неэффективными, что выявило уязвимость критически важной инфраструктуры во время повышенного беспокойства по поводу онлайн-безопасности. . • Отключение в 09:00 по Гринвичу продолжалось более семи часов, и в среду правительство подтвердило, что это была кибератака.
- Февраль 2015 г., китайские хакеры «нацелились на оборонные и финансовые фирмы США» после кибератаки на Forbes • Американские фирмы, занимающиеся кибербезопасностью, заявляют, что китайская шпионская группа взломала журнал Forbes, чтобы затем атаковать оборонных подрядчиков, финансовые фирмы и другую ничего не подозревающую добычу, посещающую популярный новостной веб-сайт. • Invincea и iSight Partners подробно рассказали о том, что они назвали кампанией «водопой» в конце прошлого года, в которой использовались преимущества Forbes.com и других законных веб-сайтов. • «Китайская продвинутая постоянная угроза скомпрометировала Forbes.com, чтобы организовать в конце ноября 2014 года атаку через Интернет против американских оборонных и финансовых компаний», — говорится в отчете Invincea, размещенном на ее веб-сайте. • «Наглая атака» использовала уязвимости в программном обеспечении Adobe Flash и Internet Explorer, которые, по данным Invincea, с тех пор были закрыты.
- 13 февраля 2015 г. Группа здравоохранения штата Теннесси уведомляет сотрудников о нарушении платежных ведомостей • Компания Franklin Healthcare Associates (SoFHA) из штата Теннесси уведомила всех сотрудников о том, что их личная информация была получена во время нарушения безопасности у стороннего поставщика платежных ведомостей компании, а некоторые уже использовался для подачи мошеннических налоговых деклараций. • Сколько жертв? Все сотрудники уведомлены, пострадали от 20 до 25 человек. • Какой тип личной информации? Информация о заработной плате сотрудников, включая формы W-2. • Что случилось? Был взломан сторонний поставщик платежных ведомостей SoFHA, был получен доступ к информации о заработной плате сотрудников SoFHA, и были поданы мошеннические налоговые декларации. • Какой был ответ? SoFHA работает с национальными, государственными и местными правоохранительными органами для выявления преступников. SoFHA уведомляет всех сотрудников и предлагает им бесплатный год услуг по защите от кражи личных данных. • Подробности: SoFHA уведомила местные органы власти в начале февраля. По состоянию на четверг от 20 до 25 сотрудников сообщили о том, что стали жертвами кражи личных данных, связанных с налогами. • Цитата: «Нам известно, что кибератака касалась только информации о заработной плате сотрудников, и ни разу не были скомпрометированы какие-либо данные пациентов», — цитирует Ричарда Панека, генерального директора SoFHA. «Мошенничество заключается в том, что преступники пытаются подать и получить возврат налога до того, как подаст заявление реальный человек».
- Вопросы • Каковы основные требования для создания CSIRT? • Какой тип CSIRT потребуется? • Какие виды услуг следует предлагать? • Насколько большой должна быть группа CSIRT? • Где в организации должна располагаться группа CSIRT? • Сколько будет стоить внедрение и поддержка команды? • Каковы начальные шаги для создания CSIRT?
- План программы CSIRT для управления сценариями для каждого типа инцидентов кибербезопасности (в худшем случае не работает, как при аварийном восстановлении)
- Что необходимо • Программа реагирования на инциденты кибербезопасности — Группы реагирования на инциденты кибербезопасности — Документированная программа реагирования на инциденты кибербезопасности — Документированный план реагирования на инциденты кибербезопасности — Документированные пособия по реагированию на инциденты кибербезопасности • Оценка внутреннего контроля • Обзор политик • Анализ пробелов • Оценка рисков REWI • Помощь в оценке рисков • Обучение по вопросам безопасности • Планирование обеспечения непрерывности бизнеса и аварийного восстановления Стандарты
- • ISO 2700 (Требования) • FFIEC • PCI DSS (обработка кредитных карт) • И многие другие стандарты и рекомендации • COBIT (Структура управления и контроля в сфере ИТ) • ISO 27005 (Управление рисками информационной безопасности) • ITIL (Структура : Определение, планирование, поставка, поддержка функций ИТ для бизнеса) Поддержание
- Общие вопросы были удалены.
- ISO и информационная безопасность 27001 Требования к информационной безопасности 27002 Кодекс практики управления информационной безопасностью 27003 Руководство по внедрению системы управления информационной безопасностью 27004 Измерение информационной безопасности 27005 Управление рисками информационной безопасности 27006 Аудит требований и сертификация ISO
- На что следует обратить внимание CSIRT взаимодействуют с другими организациями Как обращаться с конфиденциальной информацией Охватывают как операционные, так и технические вопросы •Оборудование •Безопасность •Соображения относительно кадрового состава Ресурсы для вновь формирующихся и существующих групп Заинтересованные стороны •Сотрудники CSIRT •Руководители более высокого уровня •Другие лица, взаимодействующие с CSIRT
- Не только реагирование Координация обработки инцидентов, тем самым устраняя дублирование усилий Смягчение потенциально серьезных последствий серьезной проблемы, связанной с компьютерной безопасностью Включите усилия не только в отношении способности реагировать на инциденты, но и ресурсов для оповещения и информирования группы
- Различные планы похожи друг на друга CIRP План реагирования на компьютерные инциденты CSIRP План реагирования на инциденты кибербезопасности • Группа реагирования CSIRT на инциденты кибербезопасности
- Основы программы и плана Цель Объем Допущения Собственность Действия Этапы Структура
- CSIRT Подготовка к инциденту Обнаружение Прекурсоры и анализ индикаторов Декларация Реагирование Сдерживание Ликвидация Восстановление После инцидента
- Что необходимо • Программа реагирования на инциденты кибербезопасности — Документированная программа реагирования на инциденты кибербезопасности — Документированный план реагирования на инциденты кибербезопасности — Группы реагирования на инциденты кибербезопасности — Документированные пособия по реагированию на инциденты кибербезопасности • Обзор политик • Анализ пробелов • Оценка рисков REWI — Оценка внутреннего контроля • Содействие оценке рисков • Обучение по вопросам безопасности • Планирование обеспечения непрерывности бизнеса и аварийного восстановления
- Видение Определите свой избирательный округ. Кого поддерживает и обслуживает CSIRT? Определите свою миссию, цели и задачи CSIRT. Что CSIRT делает для определенного избирательного округа? Выберите услуги CSIRT для предоставления группе (или другим лицам). Как CSIRT поддерживает свою миссию? Определить организационную модель. Как CSIRT структурирована и организована? Определите необходимые ресурсы. Какой персонал, оборудование и инфраструктура необходимы для работы CSIRT? Определите свое финансирование CSIRT. Как CSIRT финансируется для ее первоначального запуска и ее долгосрочного обслуживания и роста?
- Кто должен быть в CSIRT Teams Бизнес-менеджеры. Им необходимо понимать, что такое CSIRT и как она может помочь в поддержке их бизнес-процессов. Должны быть достигнуты соглашения относительно полномочий CSIRT в отношении бизнес-систем и того, кто будет принимать решения, если критически важные бизнес-системы должны быть отключены от сети или закрыты.
- Прекращение разрешения инициирования последовательности операций
- Документация • Программа реагирования на инциденты кибербезопасности – Документированная программа реагирования на инциденты кибербезопасности – Документированный план реагирования на инциденты кибербезопасности – Группы реагирования на инциденты кибербезопасности – Документированные пособия по реагированию на инциденты кибербезопасности Обзор политики
- • Рекомендации по использованию компьютера • Заявление о допустимом использовании • Особая политика доступа • Соглашение об особых рекомендациях по доступу • Политика сетевых подключений • Процедуры эскалации инцидентов безопасности • Процедура обработки инцидентов • Допустимое шифрование • Политика политики безопасности аналоговых/ISDN-линий • Политика безопасности лаборатории DMZ • Руководство по антивирусному процессу • Политика поставщиков услуг приложений (ASP) • Политика оценки приобретения • Стандарты безопасности ASP • Политика аудита • Политика автоматической переадресации электронной почты • Политика паролей БД • Политика коммутируемого доступа • Политика оборудования DMZ в Интернете • Политика экстрасети • Политика конфиденциальности информации • Политика безопасности внутренней лаборатории
- Обучение по вопросам безопасности имеет важное значение.